
Abstract
The fragmentation of normal alkanes during the cracking of oil under geochemical conditions with high thermal stress is important for the preservation of crude oils in reservoirs and the formation of gaseous hydrocarbons. In this study, the kinetics behavior, cracking mechanisms, and products of n-tetradecane pyrolysis were investigated using reactive molecular dynamics (ReaxFF MD) simulations at high temperatures (2000K∼3000 K). The pyrolysis process itself, its main products and intermediates, and kinetic behavior were analyzed at an atomic/molecular level. Low molecular weight (C1–C5) alkanes and olefins, together with H2, were the predominant intermediates and products of the simulations. Three distinct stages—a stable stage, initial decomposition, and secondary pyrolysis—can be distinguished during the decomposition process. The reactant n-tetradecane was exhausted when the simulation temperature reaches ∼2250 K, accompanied by a rapid increase in hydrocarbons with molecular numbers C1–C5. The yield of C2–C5 peaked at ∼2500 K and then decreased. The number of CH4 molecules increased continuously throughout the entire simulation process because of the contribution of secondary pyrolysis. The Arrhenius parameters obtained from ReaxFF MD simulations, on the basis of first-order kinetic analysis of n-tetradecane, were reasonably consistent with experimental data and generally in agreement with results from pyrolysis experiments on crude oils in the laboratory. The pyrolysis process and reaction mechanism of n-tetradecane were also reasonably consistent with laboratory pyrolysis experiments on whole oils and their individual components. ReaxFF molecular dynamics simulation is therefore considered to be a valid approach for the study of thermal cracking of subsurface crude oils.
Introduction
The investigation of pyrolysis kinetics, cracking mechanisms, intermediates, and products is crucial to understanding the thermal stability of liquid hydrocarbons and the formation of natural gas pools by oil cracking under geological conditions. Numerous oil cracking experiments have been conducted under a range of laboratory conditions to determine how the chemical compositions of oils change with increased thermal stress (Behar and Pelet, 1988; Bjorøy et al., 1988; Ungerer et al., 1988; Behar et al., 1992; Horsfield et al., 1992; Pepper and Dodd, 1995; Schenk et al., 1997; Hill et al., 2003; Tian et al., 2006, 2009; Zhao et al., 2008; He et al., 2018, 2019; Hou et al., 2022). Previous studies have reached a general understanding of the cracking process of oil. It has been generally accepted that the cracking of HMW (high molecular weight) compounds to LMW (low molecular weight) compounds is the predominant process during oil cracking, along with the formation of pyrobitumen. The generation of natural gas components (C1–C5) from oil cracking can be divided into two stages: a primary stage and a secondary stage. Gas generated during primary decomposition of liquid oil is characterized by a predominance of C2–C5 components, while secondary cracking yields methane and carbon-rich matter (Tian et al., 2006, 2009). Studies have also found that branched and cyclic compounds react or crack faster than n-alkanes and that the residual oil becomes more aromatic (Tian et al., 2006, 2009).
Raw experimental data obtained from laboratory pyrolysis are generally consistent with the theoretical model (Ungerer et al., 1988). However, the usual geochemical modeling approach using the first-order rate laws to describe the thermal decomposition of reservoir oils does not account for some observations, such as the discovery of liquid hydrocarbons in higher temperature reservoirs. Several theoretical geochemical models have therefore been developed to attempt to resolve the discrepancies between theoretical model calculations and geological cases (Ungerer et al., 1988; Dominé et al., 1990; Dominé and Enguehard, 1992; Pepper and Dodd, 1995; Waples, 2000; Behar et al., 2008). For example, Dominé and Enguehard (1992) found that the apparent kinetic parameters vary with temperature, with the activation energy of pyrolysis for hexane increasing from 65 kcal/mol at 350–400 °C to about 70 kcal/mol at temperatures below 200 °C. Dominé et al. (1998), using a simplified mechanism for hexane pyrolysis, proposed that the reaction order of hydrocarbon thermal degradation changed from 0.5 below 200 °C to 1.5 at higher temperatures. Dominé et al. (2002) inserted more free radical reactions, more chemical structures, and suitable rate constants into their geochemical models and concluded that oils can remain stable at much higher temperatures, enhancing the lower limit of stability for mature oils.
Pyrolysis is a very complicated process which usually involves a huge amount of species and chemical reactions (Battin-Leclerc, 2008). Current experimental and theoretical approaches are unable to fully describe complex pyrolysis processes and plenty of short-life intermediates produced. It is also impractical to perform direct investigation on cracking processes in crude oil, which involve many thousands of compounds.
Molecular simulation, on the other hand, can provide comprehensive insights into the intermediate processes and kinetics behavior of chemical reactions at the atomic level. This approach provides effective verification and in-depth understanding of the results of experimental and theoretical studies. In studies during the past two decades, reactive molecular dynamics (RMD) simulations using ReaxFF (van Duin et al., 2001) have been widely performed to investigate the pyrolysis of individual components of fossil fuels or petroleum (Ding et al., 2013; Castro-Marcano et al., 2014; Arvelos et al., 2019; Hong et al., 2019; Wang et al., 2021).
For instance, Ding et al. (2013) and Li et al. (2013) investigated the pyrolysis chemistry of n-heptane (C7H16) at high temperatures using RMD. Wang et al. (2011), Chen et al. (2017), and Li et al. (2021) intensively analyzed the intermediate mechanisms, product distributions, and reaction kinetics of n-dodecane (C12H26), n-hexadecane (C16H33), and n-eicosane (C20H42), respectively. The results of these studies indicate that radical reactions predominate throughout the entire pyrolysis process of normal alkanes. The initial mechanism of the pyrolysis of n-heptane, n-dodecane, or n-hexadecane is either unimolecular cleavage of C–C bonds to form smaller radicals, or dehydrogenation to produce an H radical and the corresponding n-C7H15, n-C12H25, and n-C16H33 radicals. The main product types are small fragments (C1-C3), including long-chain alkyl free radicals, small free radicals, stable products, and high-unsaturated intermediates.
The apparent activation energy for these normal alkanes extracted from the simulations is reasonably consistent with experimental results. The pyrolysis chemistries of some branched-chain alkanes—for example isobutane, isopentane, and isohexane—have also been simulated using Reax-FF based molecular dynamics and DFT theory (Xin et al., 2020). The results show that thermal decomposition occurs more rapidly in branched-chain hydrocarbons than in straight-chain hydrocarbons. The reaction rates of chain hydrocarbons with similar structures gradually increase with increase of the carbon atom number.
For naphthenic hydrocarbons, Xin et al. (2020) found that the decomposition rate of cyclopentane is slightly faster than that of cyclohexane. Due to the lower bond dissociation energy of the C–C bond in cyclobutane, the pyrolysis rate of this naphthenic hydrocarbon is significantly faster than those of other naphthenes. Liu et al. (2018) observed that homolysis of methylcyclohexane (C7H14) initiates the decomposition process, leading to opening of the ring and formation of C7H14 biradicals.
RMD pyrolysis of p-menthane (C10H20) shows that dissociation of isopropyl and the ring-opening reaction of the bond adjacent to the isopropyl are the two predominant initiations of p-menthane decomposition (Wang et al., 2021). Therefore, the type and number of substituents on rings has a significant influence on the site of bond cleavage and, subsequently, the intermediates and products.
The thermal decomposition rates of aromatic hydrocarbons are significantly lower than those of alkanes or naphthenes with similar carbon numbers because of the strong stability of the large π bond in the benzene ring (Xin et al., 2020). The pyrolysis rates of methylated aromatics are more rapid than that of their corresponding parent aromatic ring because the H atom is easily removed from CH3 group. However, most recent RMD studies on hydrocarbon pyrolysis mainly focus on the chemical kinetic mechanisms of reactions and have limited significance for guiding oil and gas exploration since the lack of combination with the geological background.
In this study, taking n-tetradecane as an example, a series of ReaxFF MD simulations was performed at temperatures ranging from 2000 K to 3000 K to investigate the effects of simulation temperatures, product distributions, and kinetics behavior on the pyrolysis process of normal alkanes. In particular, this work incorporates the ReaxFF MD simulation into a geochemical model. The results are more conducive to understanding the thermal decomposition processes of oil and the generation of natural gas under geological conditions. The apparent activation energy of n-tetradecane pyrolysis was also calculated to test the accuracy and reliability of the simulation. This study will have practical implications for informing the exploration of ultradeep buried petroleum reservoirs under high formation temperatures.
Computational method
The construction of n-tetradecane molecular models
Fifty molecules of n-tetradecane were self-assembled into a cubic box using the MS (Materials Studio software) Amorphous Cell module, as shown in Figure 1. Periodic boundary conditions in all three directions were specified for the initial cell of n-tetradecane. In order to avoid overlapping of molecules in the constructed cell, a low bulk density of 0.30 g/cm3 was taken into account. To achieve the true density of a n-tetradecane cell, a series of molecular dynamics (MD) simulations were performed using the MS Forcite module. This involved three steps:
Geometric optimization of the n-tetradecane cell was performed to ensure an accurate structure. After minimization, an anneal dynamics calculation was performed to find different local minimum energy structures of the n-tetradecane cell using the constant-pressure, constant-temperature ensemble (NPT) at temperatures from 298.15 to 700 K. The true density of the n-tetradecane cell was obtained by performing a series of MD calculations with the NPT ensemble at 298.15 K and 0.10 MPa.
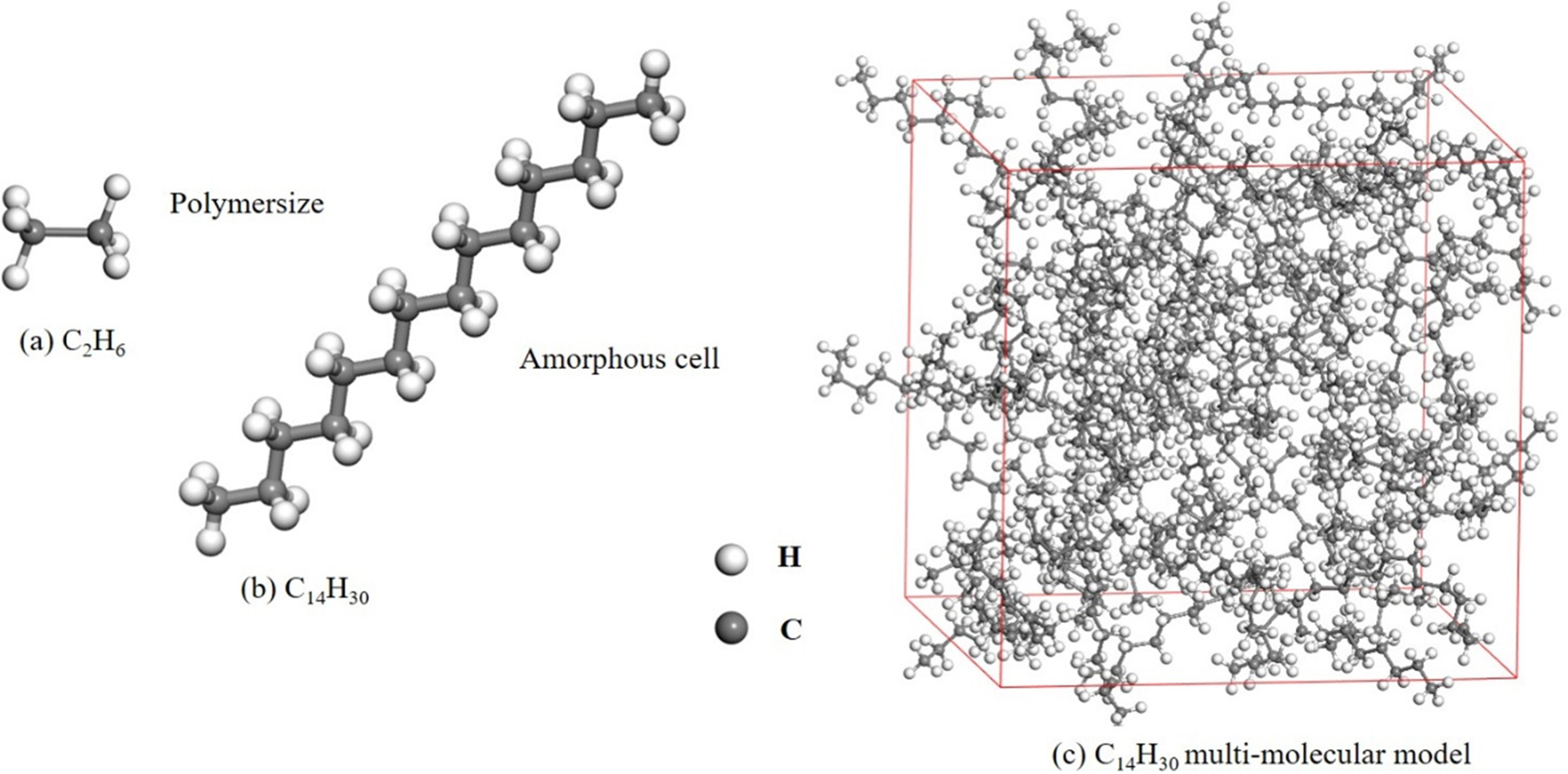
Molecular model n-tetradecane (C14H30).
The total MD simulation time was specified as 2000ps. As shown in Figure 2 (a), the equilibrium temperature of the n-tetradecane cell is equivalent to an initial temperature of 298.15 K. The density of the n-tetradecane cell is such that it should reach equilibrium at 2000ps, as shown in Figure 2 (b). The cell parameters of the n-tetradecane cell are a = 27.12 Å, b = 31.15 Å, c = 25.67 Å, α = 92.53°, β = 87.40° and γ = 82.55°. It was found that the equilibrium density was 0.764 g/cm3, which is consistent with the experimental value of 0.7594 g/cm3 (Santos et al., 2017). Both the temperature and density have reached equilibrium, which indicates that the interactions between the molecules of n-tetradecane have reached equilibrium, so the constructed model is reliable.
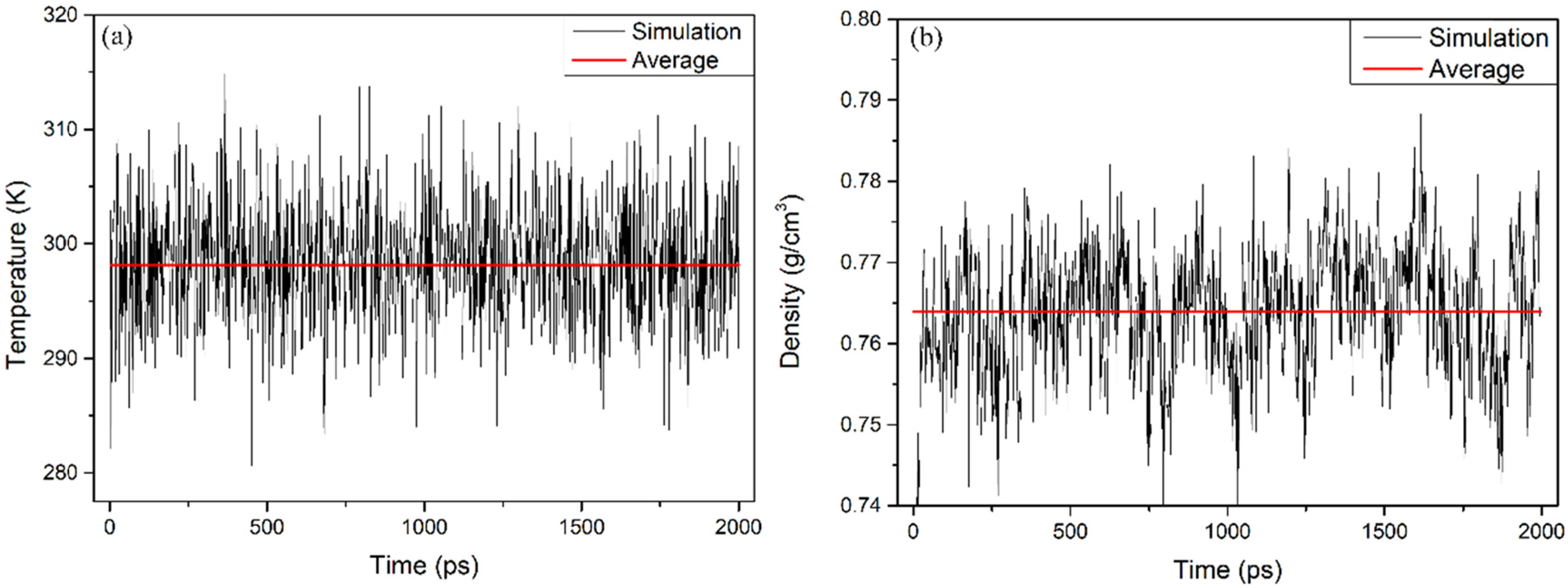
Equilibrium of temperature (a) and density (b) of n-tetradecane cell.
Reactive molecular dynamics simulation details
ReaxFF reactive force fields (Tersoff, 1986; Brenner, 1990; van Duin et al., 2001; Senftle et al., 2016) have been widely applied to obtain insights into the mechanisms of thermal pyrolysis of various hydrocarbon fuels, as they can predict bond formation and charge transfers. All thermal MD simulations in this study were performed using LAMMPS code (Aktulga et al., 2012) with ReaxFF reactive force fields. The potentials parametrized for the CH system were adopted from the work of previous work (Wang et al., 2021). To reveal the reaction mechanisms of n-tetradecane pyrolysis, a range of ReaxFF MD simulations were performed with constant temperature and volume ensembles (NVT). The two key parameters of pressure and temperature were controlled using Nose–Hoover barostat and thermostat methods (Xue et al., 2017). NVT MD simulations were then performed in a temperature range of 2000 K to 3000 K. A time step of 0.1 fs, a total simulation time of 500 ps, and a heating rate of 10 K/ps were set. The earlier decay of n-tetradecane and the subsequent formation of smaller molecules were described by a series of FORTRAN scripts (Xue et al., 2017).
Results and discussion
Effect of temperature on the process of n-tetradecane pyrolysis
To understand the effect of temperature on the pyrolysis of n-tetradecane, a series of ReaxFF MD simulations were performed on n-tetradecane in the temperature range 2000∼3000 K at intervals of 250 K. The variation in the number of n-tetradecane during the simulation process at different temperatures was shown in Figure 3. The results indicate that the decomposition rate of n-tetradecane increases dramatically with increase in temperature. At a simulation temperature of 2000 K, only 17 of the 50 n-tetradecane molecules in the initial system decomposed during the entire simulation. It can be observed that a substantial reaction occurred after 100 ps, and that the number of n-tetradecane molecules decreased steadily with simulation time. When the simulation temperature was raised to 2250 K, n-tetradecane decomposed rapidly, with the number of molecules decreasing significantly until it was completely consumed at around 400 ps. With further increases in temperature, from 2500 K to 3000 K, the rapidity of n-tetradecane decomposition increased dramatically and the number of molecules decreased abruptly.
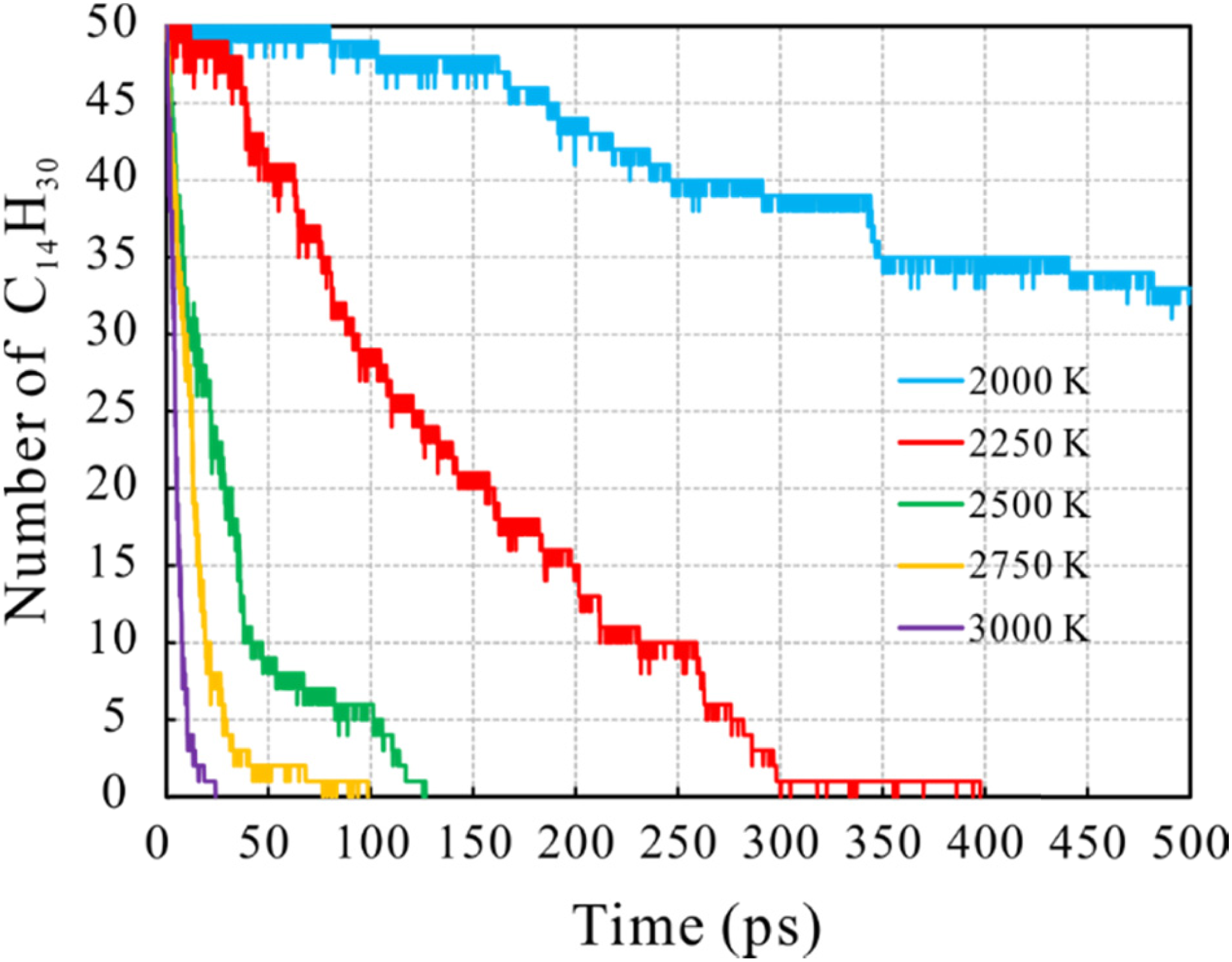
The changing of the number of n-tetradecane (C14H30) with simulation time under different simulation temperatures.
The effect of temperature on the decomposition rate during the cracking of n-tetradecane is shown in Figure 4. As seen in Figure 4, disintegration of n-tetradecane initially occurred at 79.9 ps at 2000 K, 11.9 ps at 2250 K, 0.4 ps at 2500 K, 0.3 ps at 2750 K, and 0.2 ps at 3000 K. After decomposition reaction, n-tetradecane molecules were exhausted at 397.6, 126.8, 98.6, and 24.1 ps at 2250, 2500, 2750, and 3500 K, respectively.
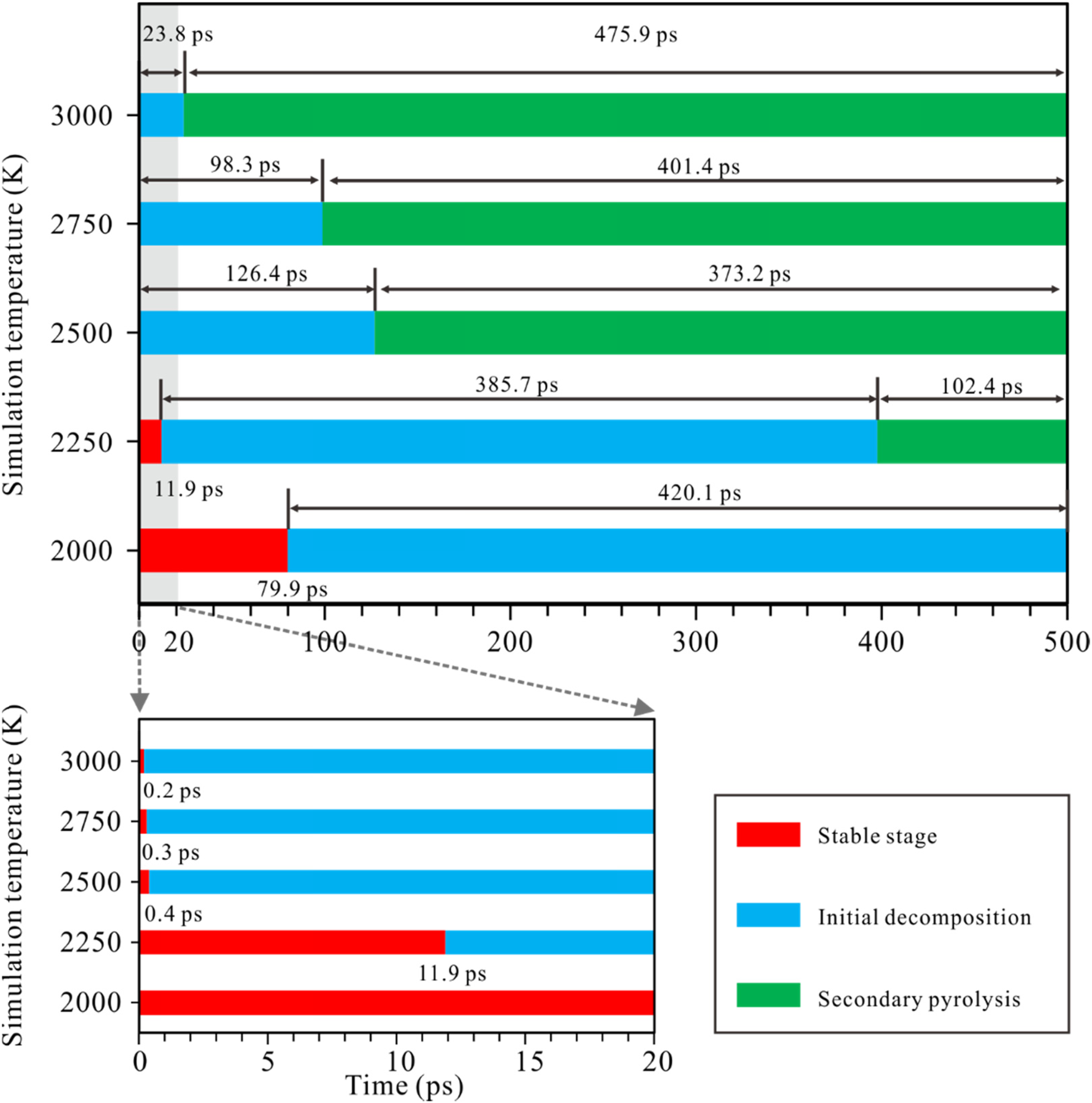
Variations of decomposition time for n-tetradecane (C14H30) ReaxFF-MD pyrolysis with the increase of simulation temperature.
The cracking process of n-tetradecane can therefore be generally divided into three stages: a stable stage, initial decomposition, and a secondary pyrolysis stage. The process in which the n-tetradecane molecules are not consumed is considered to be the stable stage. In the initial decomposition stage, the decomposition of n-tetradecane molecules continues until they are completely consumed. The secondary pyrolysis stage consists of further cleavage of the fragments produced during the initial decomposition stage.
The duration of the stable stage of n-tetradecane is radically shortened as temperature increases. Once the temperature exceeds 2250 K, n-tetradecane decomposes extremely rapidly. At simulation temperatures of 2250 K to 3000 K, the duration of the initial decomposition stage gradually decreases and secondary cracking predominates.
The three stages in the simulation generally correspond to the three stages in thermal cracking of subsurface oil to gases under geological conditions: thermal stability, primary generation of gases from liquid oil, and secondary alteration of primary gases to dry gas.
The results indicate that the onset of oil cracking corresponds to a simulation temperature of approximately 2250 K, at which point the cracking of liquid hydrocarbons with high molecular weights to light oil and condensate oil/gas is predominant. At a simulated temperature of 2500 K, the primary formation of gases from liquid oil reaches a peak. At temperatures above 2500 K, the heavy gaseous hydrocarbons (C2-5) further crack to form methane due to the increasing thermal stress.
The effect of temperature on the simulation times for the number of the total fragments, ethylene (C2H4), methane (CH4), and hydrogen (H2) was also analyzed (Figure 5). The number of total fragments increases rapidly with increased simulation temperature, indicating that increased temperature accelerates changes in the degree of pyrolysis of n-tetradecane.
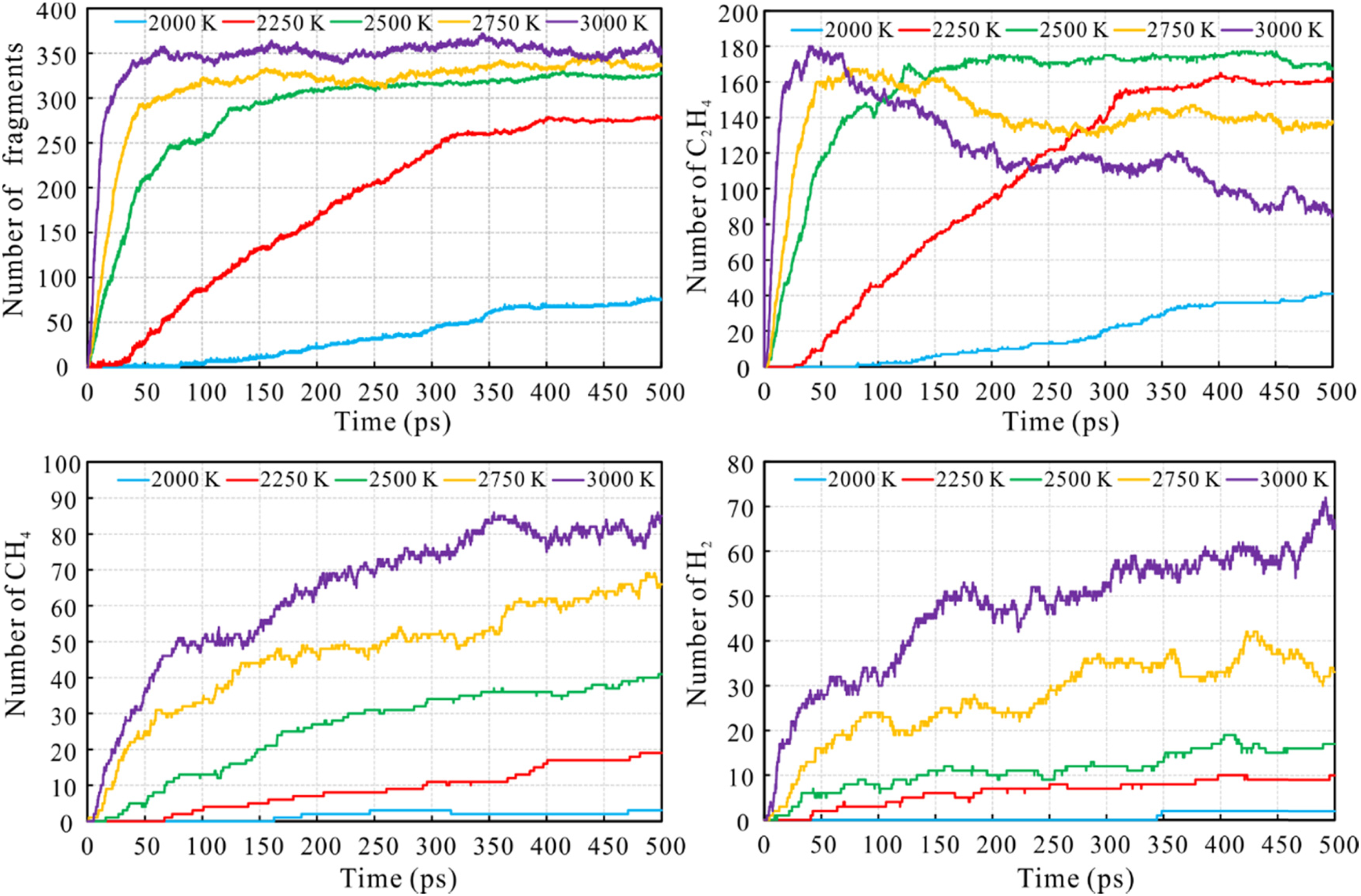
The number of (a) total fragments, (b) ethylene, (c) methane, and (d) hydrogen with simulation time during n-tetradecane ReaxFF-MD pyrolysis under different simulation temperatures.
In the simulation, ethylene is the most abundant product of pyrolysis. The number of ethylene molecules increased continuously at the beginning of pyrolysis and then gradually stabilized. However, under higher simulation temperatures, the number of C2H4 molecules increased rapidly, reaching a maximum of 168 at 2750 K and 180 at 3000 K. The C2H4 molecules then further decomposed and were consumed as the pyrolysis simulation progressed, with the rate of decomposition being substantially faster at 3000 K than at 2750 K. At the same time, the amount of methane and hydrogen generated continued to increase (Figure 5(c) and (d)), which inferred that ethylene was further decomposed to form methane through C—C bond cleavage.
In order to obtain a more visual understanding of generation of molecules or species during the MD simulation results, the snapshots of the intermediate configurations for C2H4 and CH4 have been presented in Figure 6 under different simulation temperatures at 500 ps. As can be seen from Figure 6, the left column is C2H4 and the right column is CH4 as shown in ball-and-stick style. It is distinctly found that the number of ethylene molecules decreases when the simulation temperature is greater than 2750 K, whereas the number of methane molecules continues to increase, indicating that ethylene might further decompose to generate methane at higher temperature.
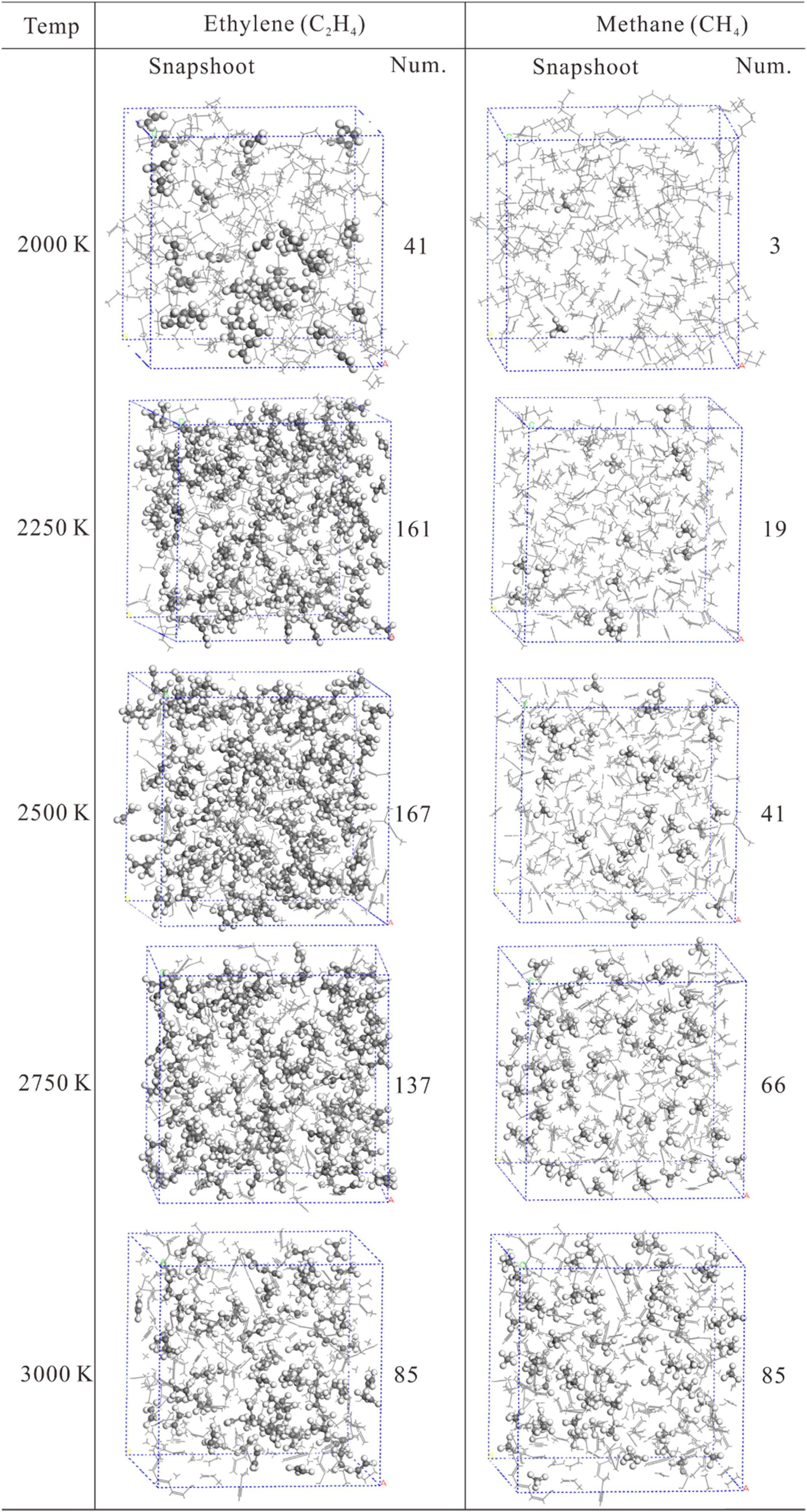
The snapshots of the intermediate configurations for C2H4 and CH4 under different simulation temperatures at 500 ps. The left column is C2H4 and the right column is CH4 as shown in ball-and-stick style.
Main species and numbers of products of n-tetradecane pyrolysis
Analysis of the number and frequency of the products of n-tetradecane pyrolysis reveals the five most abundant products at different temperatures with simulation time, as shown in Figure 7. The simulation results at 2000 K are not shown since there is barely any reaction at that temperature.
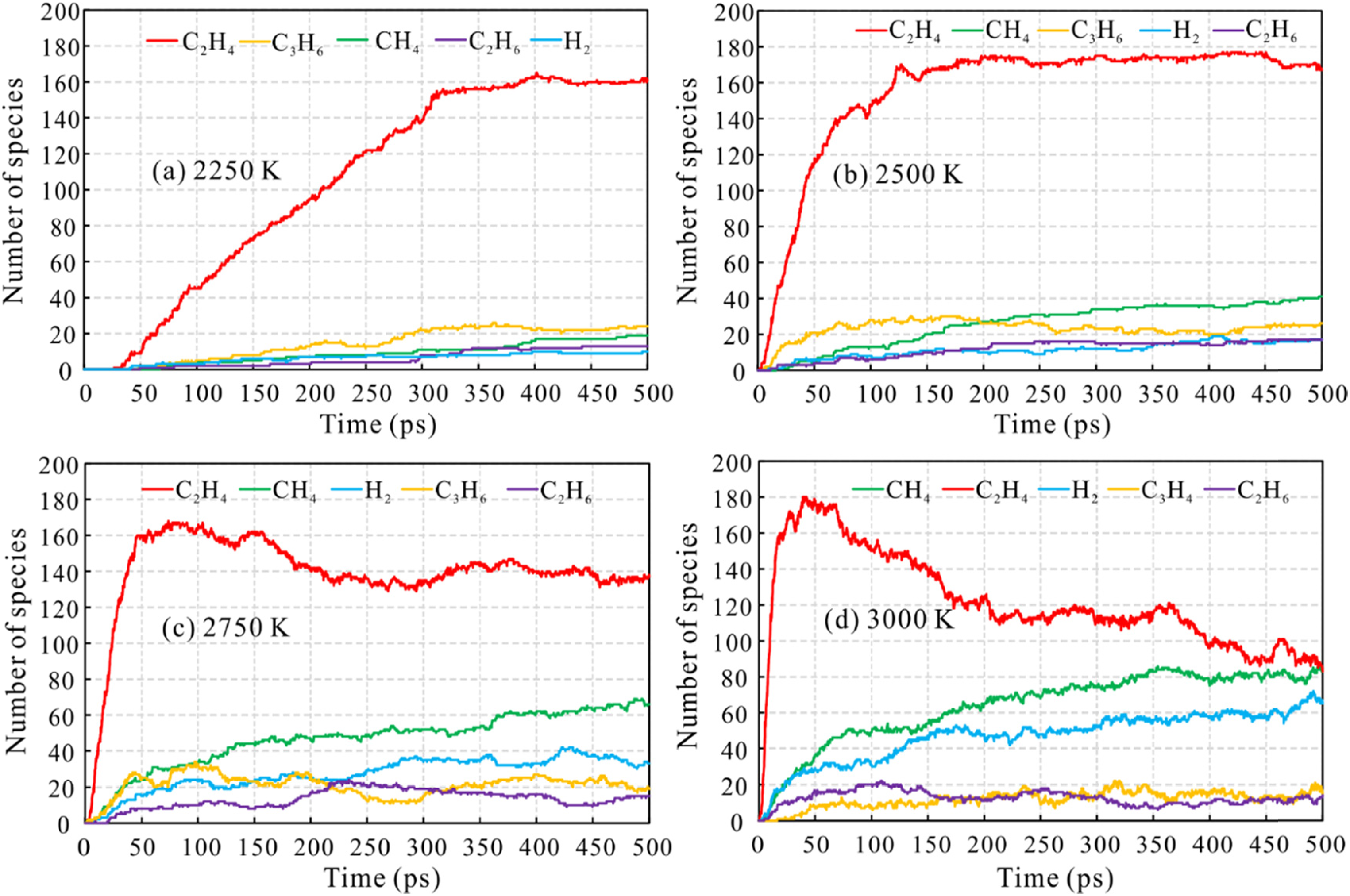
The distributions of main pyrolysis products of n-tetradecane with simulation time at different simulation temperatures: (a) 2250 K, (b) 2500 K, (c) 2750 K, and (d) 3000 K.
Ethylene (C2H4), methane (CH4), propylene (C3H6), hydrogen (H2), and ethane (C2H6) are the main products, of which C2H4 has the highest proportion and by far the most rapid production rate. At 2250 K, as the simulation time increases, the numbers of molecules of the main products continue to increase, becoming steady after about 320 ps. The numbers of molecules of other products (other than ethylene) do not exceed 25. When the temperature reaches 2500 K, the amount of CH4 keeps increasing, surpassing C3H6 (the second most abundant product). At 2750 K, the amount of C2H4 rapidly increases and then fluctuates, while the amounts of methane and hydrogen gradually increase. As the temperature ramps up to 3000 K, the pyrolysis reaction rate and degree intensify. The number of C2H4 molecules peaks sharply and then decreases in all simulation times. This is due to further decomposition of C2H4 and is in agreement with observation of n-eicosane pyrolysis (Li et al., 2013).
The numbers of molecules of n-tetradecane consumed, of the main hydrocarbon products, and of hydrogen under different temperatures at the end of 500 ps are shown in Figure 8(a). As the simulation temperature increases, the numbers of methane and hydrogen molecules increase linearly, whereas the numbers of C2H4, C2H6, and C3H6 molecules peak at 2500 K and then decrease.
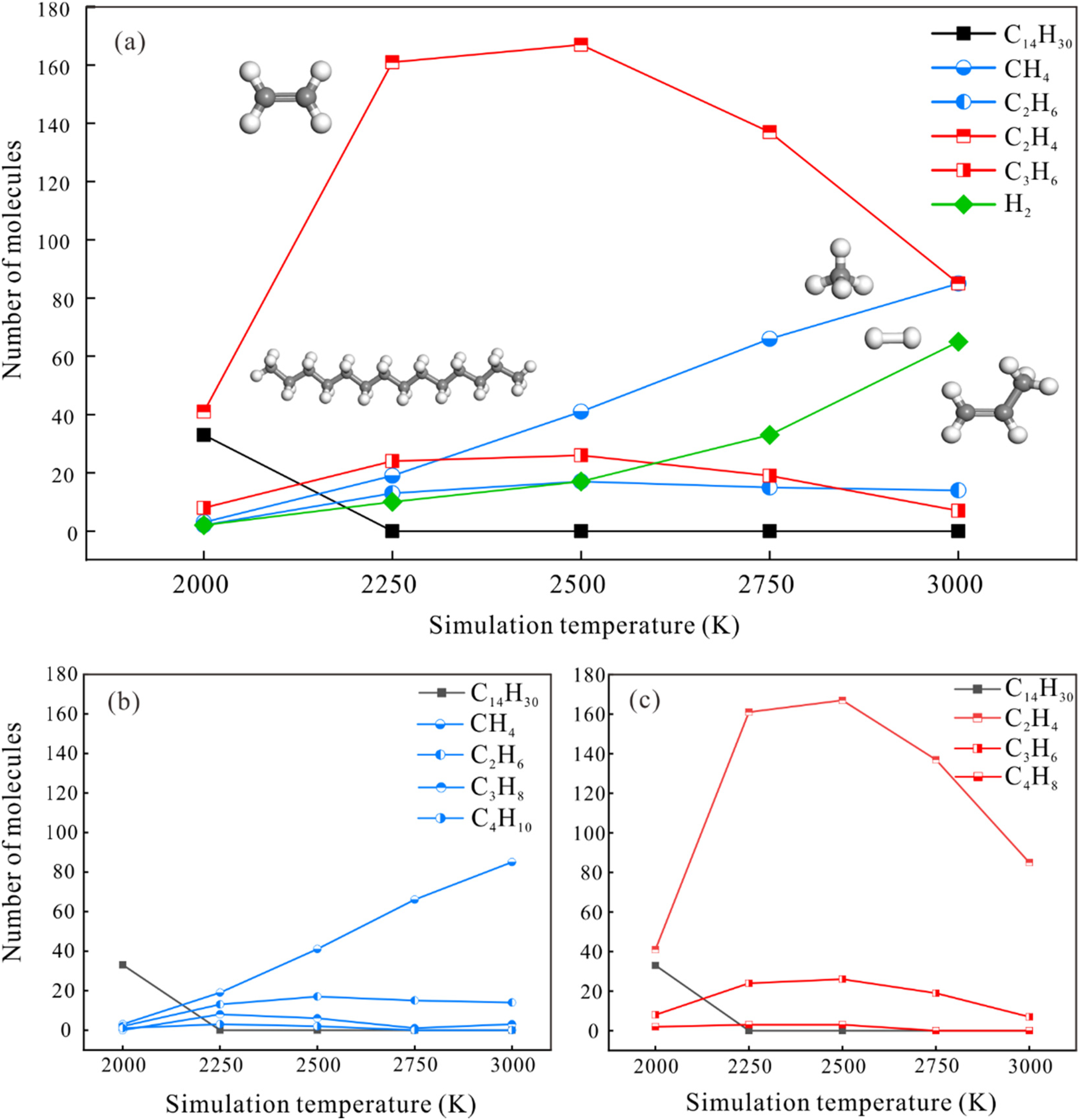
The distributions of (a) main pyrolysis products, (b) olefin products, and (c) alkane products during ReaxFF-MD pyrolysis of n-tetradecane at different simulation temperatures.
Hydrocarbon products are usually divided into two categories—alkanes and olefins—to investigate the species and characteristics of hydrocarbon products at different simulation temperatures. The numbers of alkanes and olefins at different simulation temperatures at the end of 500 ps is shown in Figure 8(b) and (c). The distribution of olefin products (C2–C4) at different temperatures has a similar trend: the numbers of C2H4, C3H6, and C4H8 molecules peak at 2500 K and then gradually decrease. However, the numbers of these homologues decrease substantially as the numbers of C atoms increases (C2–C4), with the numbers of C3H6 and C4H8 molecules produced being much lower than the number of C2H4 molecules.
Alkanes generated by the pyrolysis of n-tetradecane are mainly gaseous short-chain alkanes with low molecular weights and carbon numbers (C1–C4), i.e. CH4, C2H6, C3H8, and C4H10. Methane is the most abundant alkane during the pyrolysis of n-tetradecane, with the number of molecules increasing as the simulation temperature increases, which distinguishes it from C2H6, C3H8, and C4H10.
Methane, ethane, and propane (CH4, C2H6, and C3H8) are commonly gas components from the cracking of oil in reservoirs. Figure 9 shows the numbers of molecules and the yields (i.e. mass fraction) of these gases generated from the cracking of n-tetradecane at the end of 500 ps at different simulation temperatures. The result shows that the numbers and yields of C1–C3 alkane products have a similar evolution trend as the simulation temperature increases.
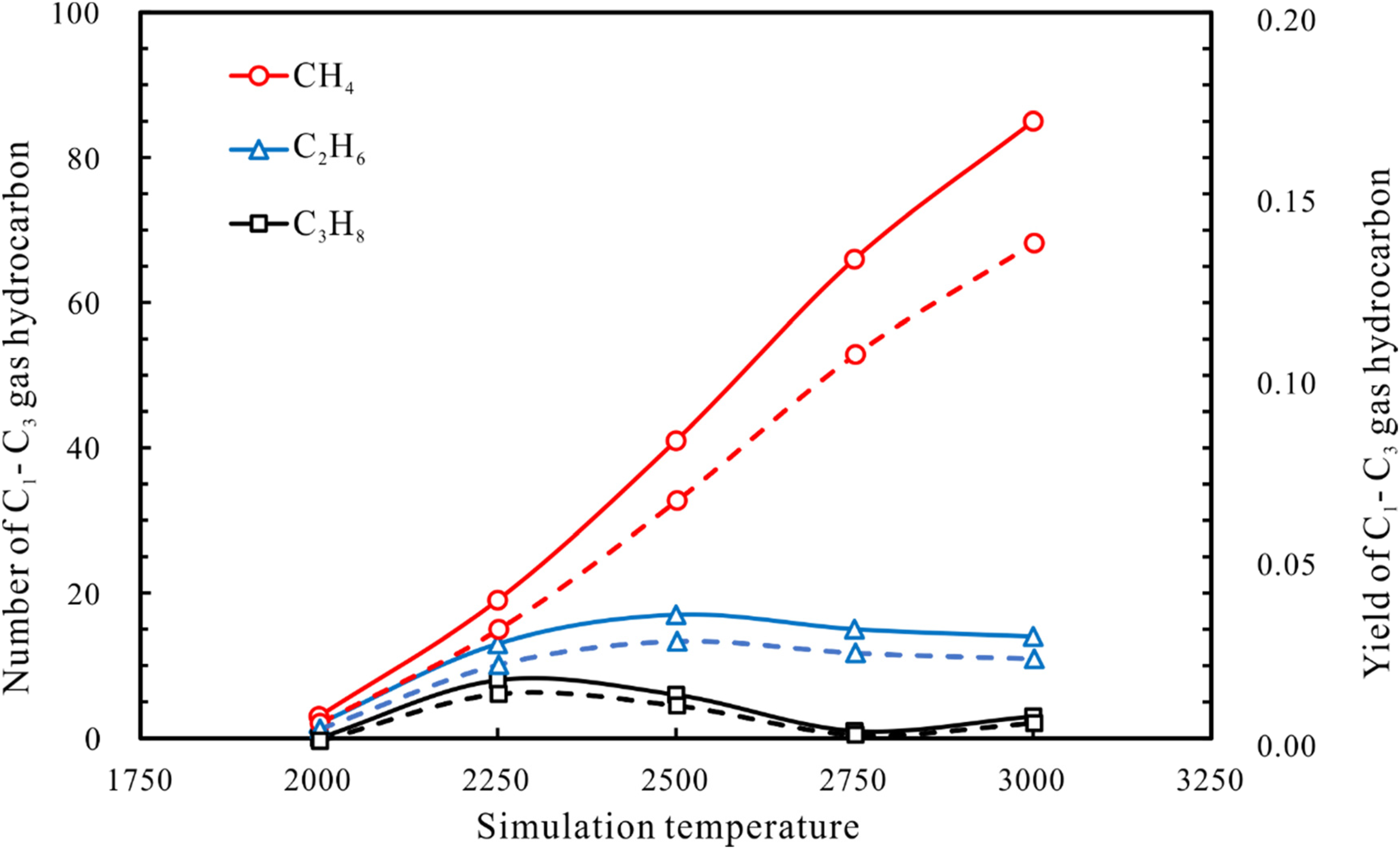
The numbers and yields of gaseous hydrocarbons (CH4, C2H6, and C3H8) generated by n-tetradecane pyrolysis.
The yield of methane is about 0.0048 at 2000 K and increases to about 0.1371 when the simulation temperature ramps up to 3000 K. The yield of C2H6 increases and then decreases as the simulation temperature increases, reaching a peak at 2500 K. The maximum yield of C3H8 occurs at a temperature lower than that of ethane, indicating that C3H8 decomposes to form methane before C2H6 does. This phenomenon is consistent with the variation in the yield of C1-C3 gaseous hydrocarbons in n-octadecane pyrolysis experiments reported by Xiong et al. (2004). Above 2500 K, the amount of generated C2–C5 short chain gaseous hydrocarbons is negligible. It can therefore be extrapolated that the secondary cracking stage of crude oil (C2-C5 gases to methane) occurs at 2500 K using ReaxFF MD simulation.
Kinetic analysis of n-tetradecane pyrolysis
The kinetic study of n-tetradecane pyrolysis is of significance for revealing the kinetic behavior and characteristics of n-alkanes in crude oil.
The consumption rate of n-tetradecane is used to describe the first-order kinetics of the thermal cracking reaction of n-tetradecane, i.e.


The Arrhenius expression was used to fit the relationship between the reaction rate and temperature, i.e.,

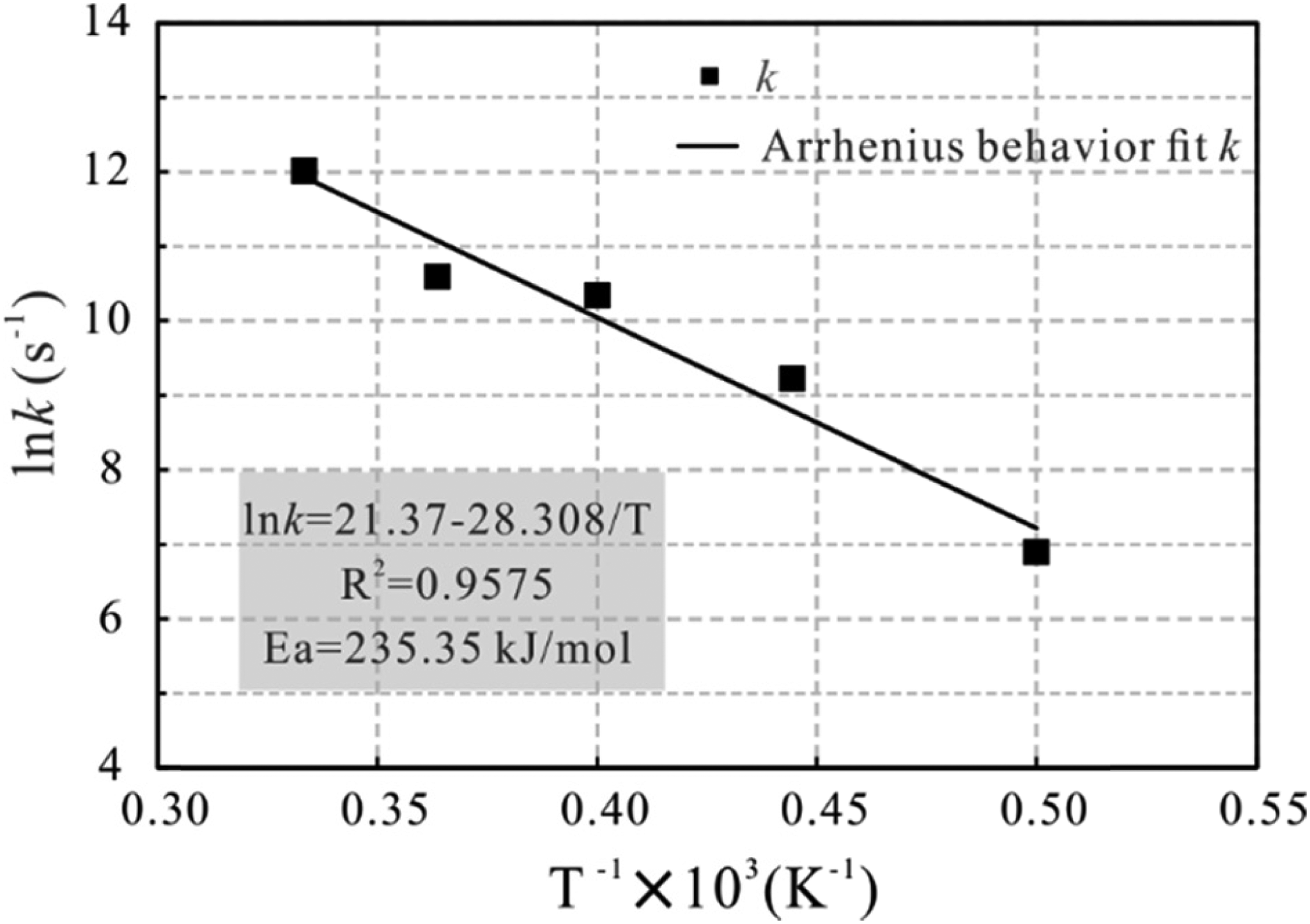
Arrhenius plot with the fitted natural logarithm of rate constants (k) versus the inverse of temperature (T). Temperature range: 2000–3000 K. Linear fit equations with R2 = 0.9575.
In addition, the kinetic parameters of n-tetradecane pyrolysis obtained from MD simulation are also consistent with previous studies on the kinetics of subsurface oil cracking in both laboratory experiments (Tian et al., 2006; Behar et al., 2008) and theoretical models (Waples, 2000): For example, Tian et al. (2006) determined the kinetic parameters for the generation of total C1–C5 gases: the mean activation energy is 249.785 kJ/mol and frequency factor A is 2.01 × 10 14 s−1. Behar et al. (2008) reported the activation energy of 57.34 kcal/mol for the disappearance of C14+ components in Boscan oil by experimental simulation in a confined system. Waples (2000) used a kinetic model to predict average activation energy of 59 kcal/mol (246.9 kJ/mol) and frequency factor of about 1.78 × 10 14 s−1 for oil destruction and gas generation under geological temperature and pressure conditions.
In general, the kinetic parameters obtained from ReaxFF MD simulations are consistent with experimental measurements of pyrolysis of both pure n-tetradecane and whole oil samples, indicating that pyrolysis of n-tetradecane can be used to describe the crude oil cracking process and that MD simulation is a practical tool for investigating kinetic behaviors of hydrocarbon pyrolysis.
Insight for crude oil cracking under geological conditions
Crude oil cracking is a very complex process involving a large number of reactions, intermediates and products (Dominé et al., 1998; Hill et al., 2003). As a common medium-chain n-alkane in the composition of crude oil, n-tetradecane is used as a representative to investigate the behaviors and characteristics of crude oil cracking process from MD simulation temperatures to geological conditions.
Figure 11 and Figure 12 show the numbers and mass yields/weight percentages of product classes observed at 500 ps at 2000K∼3000 K. As the temperature increases, the amounts and mass yields of C1–C5 and C2–C5 molecules increase rapidly, peaking at 2500 K and then decreasing slightly. In contrast, the minimum value for the variation in C6–C13 molecules corresponds exactly to the peak yield of C1–C5 and C2–C5 at 2500 K. At temperatures higher than 2500 K, the differences between C1–C5 and C2–C5 molecules in both amounts and yields gradually increase with further increase in temperature. And the number and mass yield of methane continued to increase with increasing temperature. This suggests that secondary cracking of heavy hydrocarbon gases (C2–C5 components) to generate smaller methane molecules occurs at above 2500 K. A small number of C15+ molecules (molecules with carbon numbers higher than n-tetradecane itself) were observed at 3000 K. The presence of C15+ components implies that polymerization reactions are occurring alongside n-tetradecane cracking. Xiong et al. (2004) also detected compounds with carbon numbers higher than 18 in gas chromatograms of residual liquid hydrocarbons in laboratory thermal simulation experiments on n-octadecane.
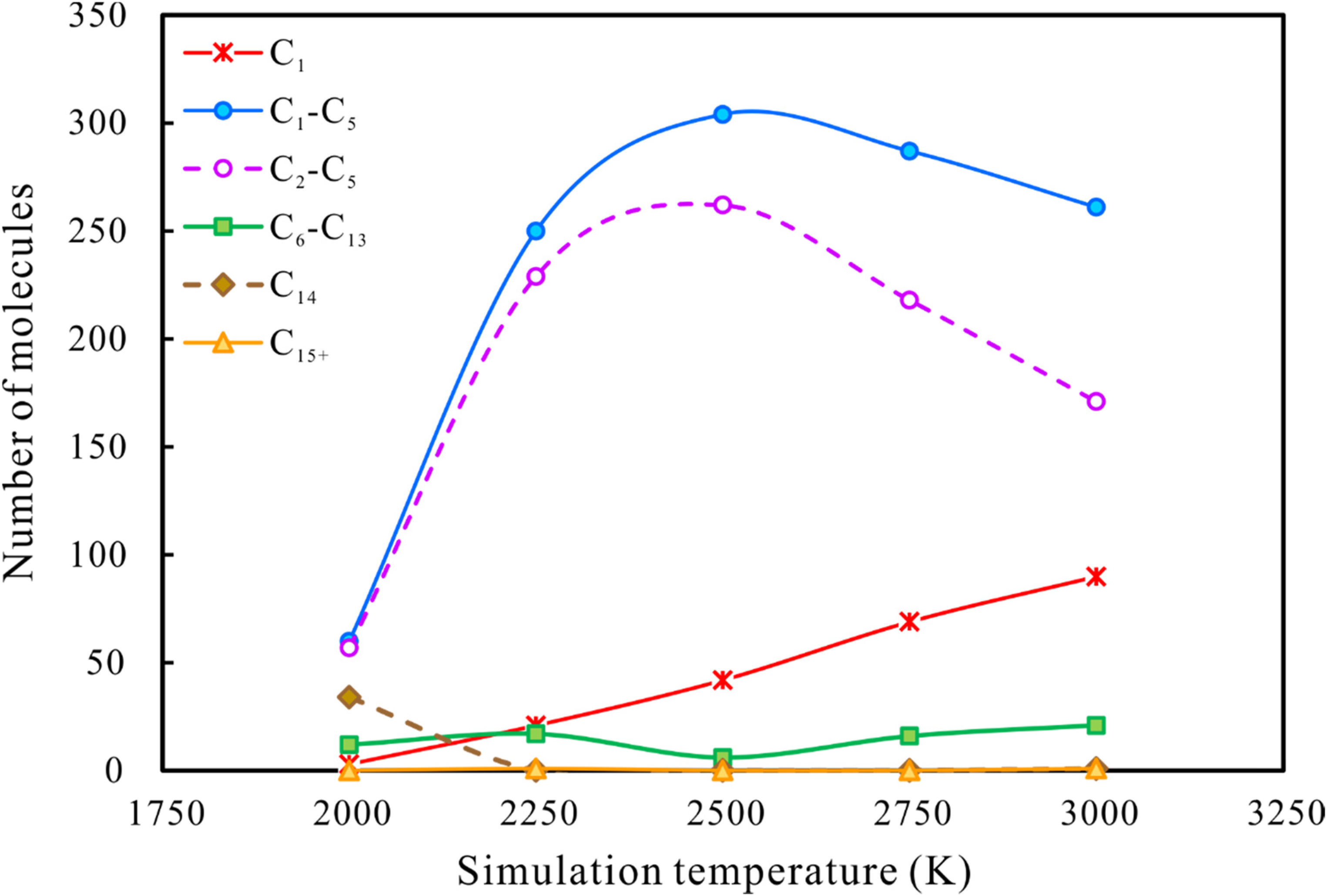
The numbers of various types of products generated by n-tetradecane pyrolysis with increasing simulation temperature.
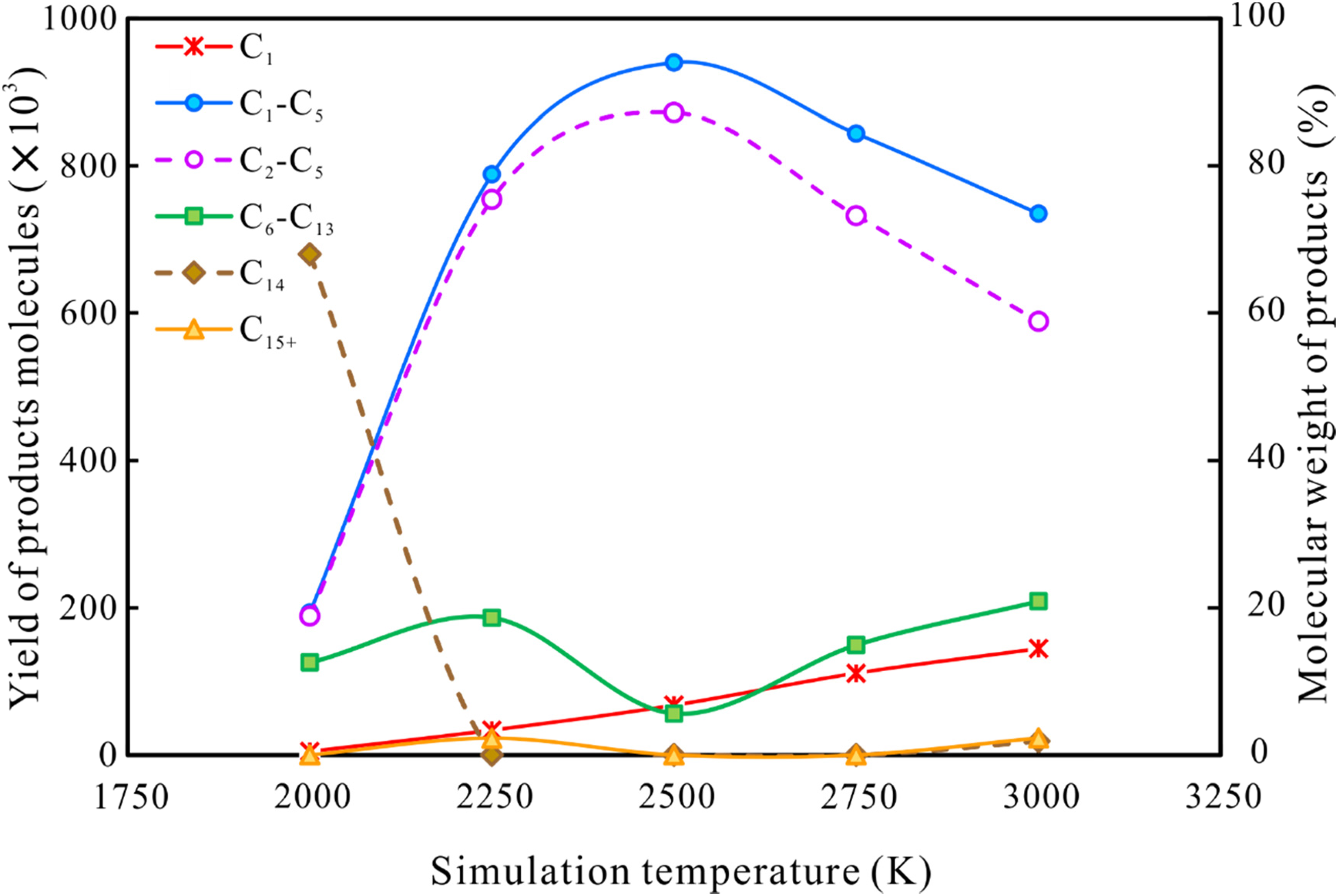
Variations of the yields (wt%) of various types of products generated by n-tetradecane pyrolysis with increasing simulation temperature.
According to the above results, the pyrolysis process of n-tetradecane can be divided into three stages, corresponding to the thermal evolution stages of crude oil (Table 1). These are: (1) thermal stability (2000K–2250 K), (2) primary formation of gases from liquid oil (2250 K–2750 K), and (3) secondary alternation of primary gases (>2750 K).
Correspondence of molecular simulation temperatures to Easy %Ro values in geological evolution of oil cracking to gas.
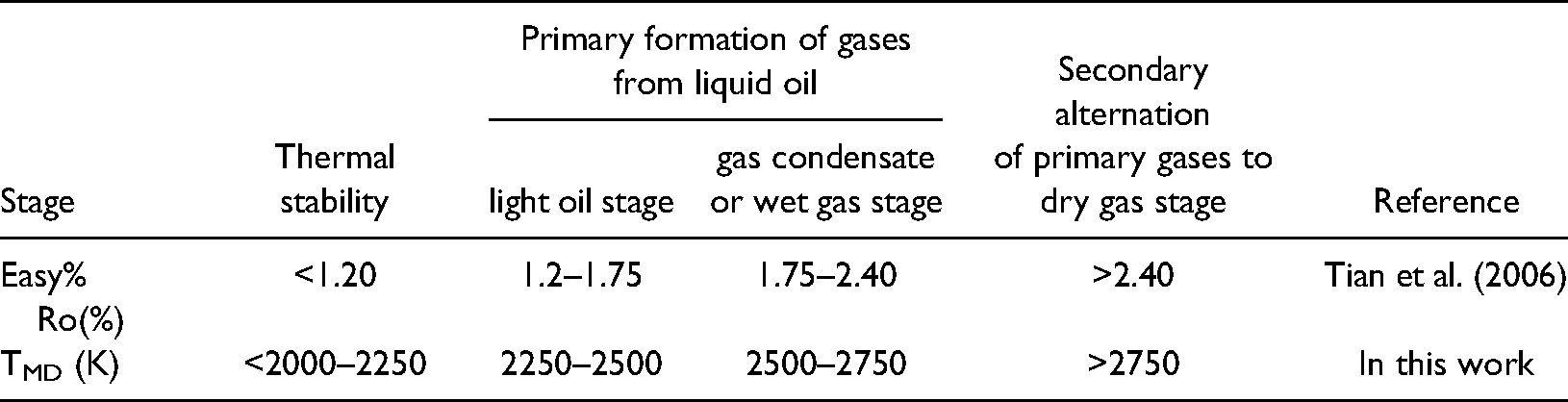
At 2000 K, crude oil remains stable without cracking. As the temperature is increased to 2500 K, crude oil begins to crack at 2250 K, corresponding to an Easy %Ro value of around 1.3%, entering the stage of primary formation of gases from liquid hydrocarbon. This is generally consistent with the accepted petroleum-geochemical paradigm that C15+ hydrocarbons are completely thermally destroyed by Ro = 1.35% (Tissot and Welte, 1984). With increased thermal stress, above a simulation temperature of 2500 K, the oil gradually becomes dominated by hydrocarbons with low molecular weights, corresponding to a value of Easy %Ro of about 1.75%. Tian et al. (2006) also reported the maximum Easy %Ro value for the preservation of separate phase oils is around 1.70%∼1.75%. When the temperature exceeds ∼2750 K on the MD time scale, wet gases crack further to form methane, indicating that the crude oil enters the stage of secondary alternation of primary gases to dry gas, corresponding to an Easy %Ro value of over 2.4% (Table 1). Therefore, the MD simulation temperature scale is reasonable and has practical implications for dividing the thermal evolution stages of crude oil cracking in this study.
ReaxFF MD simulations for hydrocarbon pyrolysis can provide new understanding for crude oil at atomic/molecular level, whereas, it is worth noting that in some circumstances the results of MD simulations are not fully comparable to those under actual geological and laboratory conditions. For example, the more intensive cleavage of C—C and C—H bonds may occur to generate considerable amounts of H2 and alkenes in MD simulations, because MD simulations shorten reaction time via increase simulation temperatures (2000K-3000 K). It is different from the observation in geological conditions (i.e. 100–300 °C), because the alkenes, as unstable compounds, may further form more stable compounds in the long geological evolution and complicated geological conditions.
Conclusions
In this paper, ReaxFF molecular dynamics is used to investigate kinetic behavior, cracking mechanisms, and products distribution during pyrolysis of n-tetradecane at different temperatures between 2000 K and 3000 K. The main intermediates and products observed in the simulations include low molecular weight (C1–C5) alkanes and olefins, together with H2. Reactant n-tetradecane is consumed at 2250 K, as the numbers of methane and hydrogen molecules continuously increase. The amount of C2–C5 hydrocarbons reaches a peak at 2500 K and then decreases owing to secondary cracking of C2–C5 gases above 2500 K. Three distinct stages are distinguished in the pyrolysis of n-tetradecane: a stable stage, initial decomposition, and secondary pyrolysis. These generally correspond to the thermal evolutionary stages of subsurface oil cracking to gas under geological conditions, viz: thermal stability, primary generation of gases from liquid oil, and secondary cracking of primary gases to dry gas. The kinetic parameters for n-tetradecane pyrolysis were obtained from the first-order reaction model ReaxFF MD simulations, which are reasonably consistent with the results from laboratory pyrolysis experiments of both pure n-tetradecane and crude oil samples. The pyrolysis process and reaction mechanism of n-tetradecane provides a reasonable insight into understanding the thermal cracking of subsurface crude oil at the atomic/molecular level. This work has promising practical implications for research on the thermal evolution of crude oil using ReaxFF MD simulations.
Footnotes
Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant No.42173054), the State Key Laboratory of Shale Oil and Gas Enrichment Mechanisms and Effective Development (Grant No.33550007-21-ZC0613-0016), and the Natural Science Foundation of Sichuan Province (No. 2022NSFSC0182). All calculated results were supported by the Sichuan University of Science & Engineering High-Performance Computing Center for providing computational. The authors thank Dr Xianggui Xue and Yushi Wen from the Institute of Chemical Materials, China Academy of Engineering Physics (CAEP) for help with the LAMMPS code.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (Grant No. 42173054), the State Key Laboratory of Shale Oil and Gas Enrichment Mechanisms and Effective Development (Grant No.33550007-21-ZC0613-0016), and the Natural Science Foundation of Sichuan Province (No. 2022NSFSC0182).