
Abstract
This narrative review comprehensively examines the multifaceted roles of proprotein convertase subtilisin/kexin type 9 (PCSK9) in peripheral arterial disease (PAD). PCSK9, a key enzymatic regulator of lipid metabolism, binds to low-density lipoprotein receptors (LDLRs) and promotes their degradation, thereby increasing plasma low-density lipoprotein cholesterol (LDL-C) levels. This mechanism establishes PCSK9 as a critical driver of atherosclerosis and PAD. Emerging evidence indicates that the role of PCSK9 in PAD extended beyond its traditional lipid-modulating functions. Specifically, PCSK9 gene polymorphisms have been shown to significantly increase PAD susceptibility, while elevated plasma PCSK9 is an independent PAD risk factor and correlates positively with disease severity. Mechanistic studies revealed that PCSK9 directly participated in vascular pathological processes through multiple pathways, some of which are independent of LDLR-mediated effects. Current clinical applications have demonstrated that PCSK9 inhibitors confer clinically meaningful therapeutic benefits in PAD patients, including reduced risks of major adverse cardiovascular events (MACE) and major adverse limb events (MALE). In particular, some of these clinical benefits may be attributed to improvements in endothelial function and microcirculatory perfusion. However, the therapeutic strategies involving PCSK9 inhibitors face persistent challenges, such as determining optimal dosing timelines, assessing long-term safety, and exploring synergies with other therapies. Future studies should prioritize large-scale randomized controlled trials to elucidate the molecular mechanisms underlying PCSK9's roles in vascular calcification and inflammatory microenvironments. Additionally, advancing the precision interventions targeting PCSK9 is essential for advancing comprehensive management approaches for PAD.
Keywords
Introduction
Proprotein convertase subtilisin/kexin type 9 (PCSK9), a predominantly liver-secreted serine protease, exerts its canonical function through binding to hepatic low-density lipoprotein receptors (LDLRs). This interaction facilitates lysosomal degradation of LDLR on hepatocyte surfaces, thereby reducing hepatic uptake of low-density lipoprotein cholesterol (LDL-C) and resulting in elevated plasma LDL-C levels.1,2 Emerging studies have further elucidated the multifaceted roles of PCSK9 in atherosclerosis: beyond driving vascular pathology via dyslipidemia, it directly contributes to vascular endothelial dysfunction, inflammatory cascades, phenotypic switching of vascular smooth muscle cells (VSMCs), and pathological vascular calcification.3–5
Peripheral arterial disease (PAD), an advanced stage of systemic atherosclerosis, affects over 200 million adults worldwide (with a 2015 systematic review estimating approximately 237 million cases globally) and is associated with significant risks of critical complications, including limb ischemia and increased cardiovascular mortality.6,7 Current first-line lipid-lowering therapy relies on statins to reduce LDL-C, a well-established driver of atherosclerosis. Over the last decade, numerous studies have confirmed the importance of lowering LDL-C in patients with established cardiovascular risk, particularly targeting this specific lipid marker.8,9 However, statin intolerance often hinders optimal lipid control due to adverse effects such as muscle pain or liver enzyme elevations. Of note, statin intolerance affects approximately 20% of patients, further impeding LDL-C management and leaving residual cardiovascular risk unaddressed. 10 Consequently, the limited availability of effective alternative treatments for this substantial patient population remains a major clinical challenge
Importantly, PCSK9 gene polymorphisms associate with PAD susceptibility. As novel lipid-lowering agents, PCSK9 inhibitors (PCSK9is; e.g. evolocumab and alirocumab) significantly reduce LDL-C levels and cardiovascular events in PAD patients. Moreover, growing evidence indicates PCSK9 regulates peripheral vascular function beyond lipid modulation.11,12 Critically, recent studies reveal PCSK9 directly exacerbates PAD progression via non-lipid mechanisms, including vascular inflammation and arterial calcification. 1 To address statin limitations, this review synthesizes PCSK9-driven pathophysiological mechanisms, evaluates therapeutic applications, and proposes future research for targeted interventions.
Currently, there is still some unclear knowledge in the pathological mechanisms and therapeutic strategies for PAD. Although PCSK9is have been recommended by clinical guidelines as an intensive lipid-lowering option for high-risk patients, the specific molecular targets, long-term efficacy, and synergistic effects with existing therapies in PAD remain to be thoroughly investigated. An elucidation of the mechanisms by which PCSK9 mediates vascular inflammation, pathological calcification, and cellular senescence will provide novel insights for developing precision interventions in PAD management. This narrative review is guided by the Scale for the Assessment of Narrative Review Articles (SANRA). 13
Genetic determinants of PCSK9 in PAD pathogenesis
PCSK9 gene polymorphisms exhibited significant associations with the risk of developing PAD. Gain-of-function mutations enhanced the ability of PCSK9 to mediate the degradation of LDLRs, leading to abnormally elevated plasma levels of LDL-C and consequently increasing the risk of atherosclerosis. 8 For instance, patients with familial hypercholesterolemia carrying specific PCSK9 mutations exhibited markedly higher plasma LDL-C levels compared to the general population, accompanied by a significantly elevated incidence of PAD within this cohort. 14
Conversely, loss-of-function mutations confer cardiovascular protection by reducing LDL-C levels. The PCSK9 genetic score (PCSK9-GS), established through a collaborative cohort study in China and the United Kingdom, revealed that a higher PCSK9-GS is significantly associated with reduced carotid plaque formation, a decreased incidence of major occlusive vascular events, and a lower risk of ischemic stroke. Significantly, certain PCSK9 genetic variants may exert dual effects: while specific variants reduce overall cardiovascular risk, they may simultaneously increase susceptibility to PAD. 15
Furthermore, PCSK9 polymorphisms significantly interact with gene–environment factors. Clinical evidence indicates that co-exposure to smoking and specific PCSK9 variants (e.g. rs505151) resulted in a synergistic increase in PAD risk compared to the presence of either risk factor alone. This gene–environment interplay provided critical insights for risk stratification in PAD. 16
Epidemiological and clinical associations of PCSK9 with PAD
PCSK9 as an independent biomarker for PAD risk
Accumulating clinical studies have demonstrated a significant association between elevated plasma PCSK9 levels and an increased risk of PAD. A case-control study involving 248 patients with intermittent claudication and 251 baseline-matched controls revealed significantly higher plasma PCSK9 concentrations in the PAD group comparing with the control group (250 ± 77 vs. 222 ± 68 ng/mL, p < 0.001). Age-adjusted logistic regression analysis indicated that every 100 ng/mL increase in plasma PCSK9 concentration was associated with a 1.78-fold elevated risk of PAD (odds ratio [OR] = 1.78, 95% confidence interval [CI]: 1.03–2.33, p < 0.001). It is worth emphasizing that, this association remained statistically significant even after further adjustment for lipoprotein(a)-adjusted LDL-C, HDL-C (high-density lipoprotein cholesterol), high-sensitivity C-reactive protein (hs-CRP), statin therapy history, and traditional cardiovascular risk factors, including hypertension, diabetes, and smoking (adjusted OR = 1.49, 95% CI: 1.03–2.18, p = 0.035). These findings suggest that PCSK9 may serve as an independent predictor of PAD risk. 10
This observation was further validated in a cross-sectional study of 115 participants (44 PAD patients vs. 71 individuals without atherosclerotic disease). The study showed markedly elevated plasma PCSK9 levels in the PAD group (471.6 ± 29.6 vs. 302.4 ± 16.1 ng/mL, p < 0.001). Multivariate linear regression analysis confirmed an independent correlation between PAD status and PCSK9 levels after controlling for confounding variables (β = 0.32, p = 0.008). 12 These two studies underscored the predictive value of circulating PCSK9 levels in assessing PAD risk through distinct methodological designs.
Correlation between PCSK9 and disease severity
Beyond disease risk, emerging evidence suggests that circulating PCSK9 levels may correlate with the clinical progression of PAD. Plasma PCSK9 levels were positively correlated with both the number of chronic total occlusion (CTO) lesions (r = 0.40, p < 0.05) and the count of affected vascular segments (r = 0.36, p < 0.05) in patients with confirmed PAD by computed tomography angiography (CTA) assessments. 12 These findings imply that PCSK9 may serve as a biomarker reflecting the severity of atherosclerotic burden.
However, heterogeneity exists among studies investigating the relationship between PCSK9 and PAD. A prospective cohort study involving 1447 community-dwelling individuals (median follow-up: 4.8 years) demonstrated no significant association between baseline plasma PCSK9 levels and carotid-femoral pulse wave velocity (cf-PWV, a measure of arterial stiffness; β = 0.02, p = 0.67 after multivariable adjustment). 17 These contradictory findings may stem from differences in study endpoints: cf-PWV predominantly reflected arterial elastic function, while PAD is fundamentally characterized by occlusive arterial pathology. This divergence suggests that PCSK9 may contribute to distinct phenotypes of atherosclerosis through heterogeneous mechanisms. Further large-scale, multicenter studies integrating molecular mechanisms are warranted to clarify the pathophysiological role of PCSK9 in PAD progression.
Mechanistic insights into PCSK9-driven PAD pathophysiology
PCSK9 normal expression and physiological functions
The PCSK9 gene is located on human chromosome 1p32.3 and encodes a protein consisting of 692–694 amino acids. This protein is highly expressed in the liver, intestine, and kidneys. In the liver, PCSK9 is primarily synthesized by hepatocytes and secreted into the bloodstream. 18 Its canonical function involves binding to the LDLR, promoting LDLR degradation, thereby reducing LDLR availability and impairing the plasma clearance of LDL-C.1,2 Additionally, PCSK9 is expressed in extrahepatic tissues, including VSMCs, macrophages, endothelial cells (ECs), pancreatic islets, renal tubules, intestinal enterocytes, and the central nervous system. 19
The expression of PCSK9 is regulated by multiple factors, including transcription factors, post-translational modifications (e.g. phosphorylation, glycosylation, and sulfation), and secretory mechanisms. For instance, PCSK9 secretion requires autocatalytic cleavage within the endoplasmic reticulum (ER) and is mediated by transport via COPII-coated vesicles. Additionally, PCSK9 is further modulated by regulatory proteins such as FAM20C 19 (Figure 1).
The canonical functions of PCSK9 include binding to the low-density lipoprotein receptor (LDLR) to promote LDLR degradation, thereby reducing LDLR availability. Additionally, PCSK9 enhances oxidized LDL (ox-LDL) uptake, activates inflammatory signaling pathways, and inhibits cholesterol efflux.
Regulation of vascular endothelial function
Molecular mechanisms
In hyperglycemic conditions and diabetic models, ECs exhibit significantly elevated PCSK9 expression in plasma and vascular tissues. Notably, in vitro studies demonstrate that PCSK9 facilitates E3 ubiquitin ligase NEDD4 binding to VEGFR2, promoting its ubiquitination and degradation. This leads to suppressed VEGFR2 signaling in ECs. Furthermore, cellular experiments indicate that PCSK9 inhibits AKT/eNOS/ERK1/2 phosphorylation under hyperglycemic stress, reducing nitric oxide (NO) bioavailability while increasing superoxide anion (O₂−) generation, thereby impairing endothelial function and angiogenesis. Critically, studies in diabetic rodent models report that evolocumab (a PCSK9 monoclonal antibody) counteracts the hyperglycemia-induced suppression of NO synthesis and attenuates O₂− overproduction. It also restores AKT/eNOS/ERK1/2 signaling, improves endothelial function, and accelerates wound healing. However, the direct translation of these therapeutic benefits to human diabetic vascular complications requires validation, given inherent differences between murine wound healing and human diabetic foot ulcers (DFUs) 20 (Figure 2).

PCSK9 upregulation in hyperglycemia promotes NEDD4-mediated VEGFR2 ubiquitination/degradation, suppresses AKT/eNOS/ERK1/2 phosphorylation, reduces NO bioavailability, and increases O2− production in endothelial cells. Abbreviations: PIP3, phosphatidylinositol 3,4,5-trisphosphate; AKT, protein kinase B; mTORC, mammalian target of rapamycin complex; Raf, rapidly accelerated fibrosarcoma; BAD, Bcl-2 associated death promoter.
Mechanistically, in vitro evidence suggests recombinant PCSK9 induces defective phagocytosis of apoptotic ECs and upregulates senescence-associated β-galactosidase (SA-β-gal) activity, while PCSK9 deficiency restores apoptotic cell clearance.17,18 PCSK9 also downregulates the anti-apoptotic Bcl-2 and upregulates the pro-apoptotic Bax, activating caspase-9/3-dependent apoptosis. Conversely, in cultured ECs, PCSK9 silencing activates autophagy under ox-LDL stress and mitigates endothelial injury by suppressing PI3 K/AKT/mTOR. 21 Whether these mechanisms operate identically in human atherosclerotic microenvironments remains unclear, necessitating studies in patient-derived cells/tissues22,23 (Figure 3).

PCSK9 silencing indeed activates autophagy induced by oxidized low-density lipoprotein (ox-LDL). This process is primarily mediated through the PI3 K/AKT/mTOR signaling pathway. Additionally, PCSK9 silencing reduces inflammatory factor release, promotes endothelial cell proliferation, and suppresses apoptosis. Abbreviations:ox-LDL, oxidized low-density lipoprotein; MCP1, monocyte chemotactic protein 1; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; CREB, cAMP-response element binding protein; Ikkα/β, inhibitor of kappa B kinase alpha/beta.
Microcirculatory dysfunction amelioration clinically
PCSK9 critically regulates microcirculatory function. Clinical studies indicated that PCSK9is significantly improve peripheral microvascular perfusion. In the aforementioned prospective trial, 6-month PCSK9i therapy in 32 patients yielded: (a) increased peripheral tissue oxygen saturation (StO2) from 67 ± 12% to 71 ± 11% (+7 ± 6%, p = 0.012); (b) enhanced flow-mediated dilation (FMD) from 5.4 ± 1.7% to 6.4 ± 1.9% (p < 0.001); (c) reduced pulse wave velocity (PWV) in male patients from 8.9 ± 2.1 m/s to 7.9 ± 1.5 m/s (p < 0.01); and (d) lowered aortic augmentation index (AIx) from 27.1 ± 10.4% to 23.0 ± 9.7% (p < 0.001). These improvements in microvascular function correlated positively with FMD changes and inversely with PWV and AIx. Notably, the effects were independent of LDL-C reduction, suggesting non-lipid pathways mediate PCSK9i-induced microcirculatory benefits. 11 This restoration likely synergized with endothelial function.
A prospective intervention study showed that 32 patients at very high cardiovascular risk were treated with a PCSK9i for 6 months. Brachial artery FMD increased significantly from a baseline of 5.4 ± 1.7% to 6.4 ± 1.9% (o + 19 ± 10%, p < 0.001). Importantly, this endothelial protective effect was independent of changes in LDL-C levels. 11 These results indicate the direct vascular benefits of PCSK9 inhibition.
Driving vascular inflammation and senescence
Pro-inflammatory mechanisms
Cytokine cascade activation. In vitro studies demonstrate that PCSK9 significantly upregulates the mRNA expression of IL-1β, TNF-α, and MCP-1 (p < 0.01) in ECs. 24 Utilizing human induced pluripotent stem cell (iPSC)-derived vascular models, the small-molecule PCSK9i NYX-1492 (which blocks the PCSK9-LDLR interaction) reduced monocyte adhesion in tissue-engineered blood vessels (TEBVs) and suppressed VCAM-1/TNF-α secretion during iPSC differentiation into vascular cells. 25 While these engineered systems provide valuable mechanistic insights, their simplified microenvironment lacks physiological hemodynamic forces and immune crosstalk. Nevertheless, these data suggest that PCSK9 inhibition may attenuate vascular inflammation independently of LDL-lowering, potentially enhancing anti-atherosclerotic effects in high-inflammation subgroups. 25
LDLR-independent inflammatory pathways. Notably, studies involving Ldlr−/− mice reveal that PCSK9 accelerates atherosclerosis through LDLR-independent mechanisms. Adenylate cyclase-associated protein 1 (CAP1) has been identified as a key mediator of PCSK9-driven inflammation, facilitating cytokine induction, TLR4 upregulation, and enhanced ox-LDL uptake. Mechanistically, spleen tyrosine kinase (Syk) and protein kinase C dependent mechanisms. Adenylate cyclase-associated protein 1 (CAP1) has been identified as a key mediator of PCSK9-driven inflammation, facilitating cytokine vely with Syk/PKCδ/p65 phosphorylation. 26 Importantly, the CAP1-Fc fusion protein demonstrated stronger suppression of inflammatory signaling in vitro than evolocumab, 26 though its in vivo efficacy and translational potential require validation in clinical settings.
Clinical correlations with systemic inflammation. Clinical evidence supports the pro-inflammatory role of PCSK9: In 401 patients with type 2 diabetes, plasma PCSK9 positively correlated with hs-CRP, fibrinogen, and leukocyte count. 27 Additionally, PCSK9 levels were associated with triglycerides, hs-CRP, sCD40L, and sP-selectin, with stronger correlations observed in diabetic subgroups. 28
These observational data indicate associations rather than causation, and confounding metabolic factors cannot be excluded. Future mechanistic studies in human vascular tissues are warranted to establish direct pathological links.
Accelerated vascular aging
Circulating PCSK9 levels positively correlate with aging and cardiovascular dysfunction in observational studies. PCSK9 is implicated as a determinant of age-related dyslipidemia and is associated with increased carotid intima-media thickness (IMT), a clinical marker of vascular aging. 29 However, the causal role of PCSK9 in human vascular aging requires verification in longitudinal cohorts. Preclinical evidence suggests therapeutic potential: In aged rat models, pharmacological inhibition of PCSK9 with alirocumab attenuated cardiovascular disease progression, 29 though translation to humans necessitates evaluation given interspecies differences in aging pathways. Proposed mechanisms from experimental models: (a) SIRT1 suppression: In vitro studies indicate PCSK9 accelerates endothelial senescence by downregulating SIRT1 (a key anti-aging deacetylase). PCSK9 inhibition restored SIRT1 levels in rodent vessels, 30 but human vascular relevance remains unconfirmed. (b) Senescence marker induction: Recombinant PCSK9 protein induced defective apoptotic clearance and upregulated senescence-associated β-galactosidase (SA-β-gal) in cultured ECs. 23 These findings require validation in human aging vasculature, where microenvironmental complexity may alter outcomes.
Mediating VSMC phenotypic switching and calcification
Phenotypic switching in atherosclerosis
Calcification and limb perfusion impairment. Under pro-calcific culture conditions, PCSK9-overexpressing human VSMCs showed increased extracellular calcium deposition (p < 0.01), with elevated calcifying extracellular vesicles. 33 This medial calcification directly impairs arterial compliance in distal limb vessels, reducing tissue perfusion reserve and exacerbating claudication severity. In diabetic PAD, PCSK9-mediated calcification may accelerate microvascular compromise, contributing to critical limb ischemia (CLI) and amputation risk. However, direct evidence linking PCSK9 specifically to human PAD-associated calcification patterns (e.g. Mönckeberg's sclerosis) remains limited by inadequate in vivo models. Although these data suggest that PCSK9 promotes vascular calcification, direct evidence linking this mechanism to human PAD calcification is lacking, particularly given the absence of in vivo hemodynamic and metabolic cues in monolayer models.
Hemodynamic modulation in peripheral arteries. PCSK9 overexpression exacerbated homocysteine-induced phenotypic switching in VSMCs, 34 particularly relevant in PAD patients with hyperhomocysteinemia. The mechanosensitive regulation suggests PCSK9 drives site-specific VSMC dysfunction in peripheral arterial beds, warranting investigation in human PAD specimens with disturbed flow.
Promoting multifaceted progression of atherosclerosis
Elevated levels of PCSK9 are clinically associated with coronary, cerebrovascular, and peripheral atherosclerotic diseases, though mechanistic causality requires further validation.
PCSK9-mediated macrophage dysfunction in atherogenesis
Experimental evidence indicates that PCSK9 contributes to atherosclerosis by disrupting macrophage homeostasis.
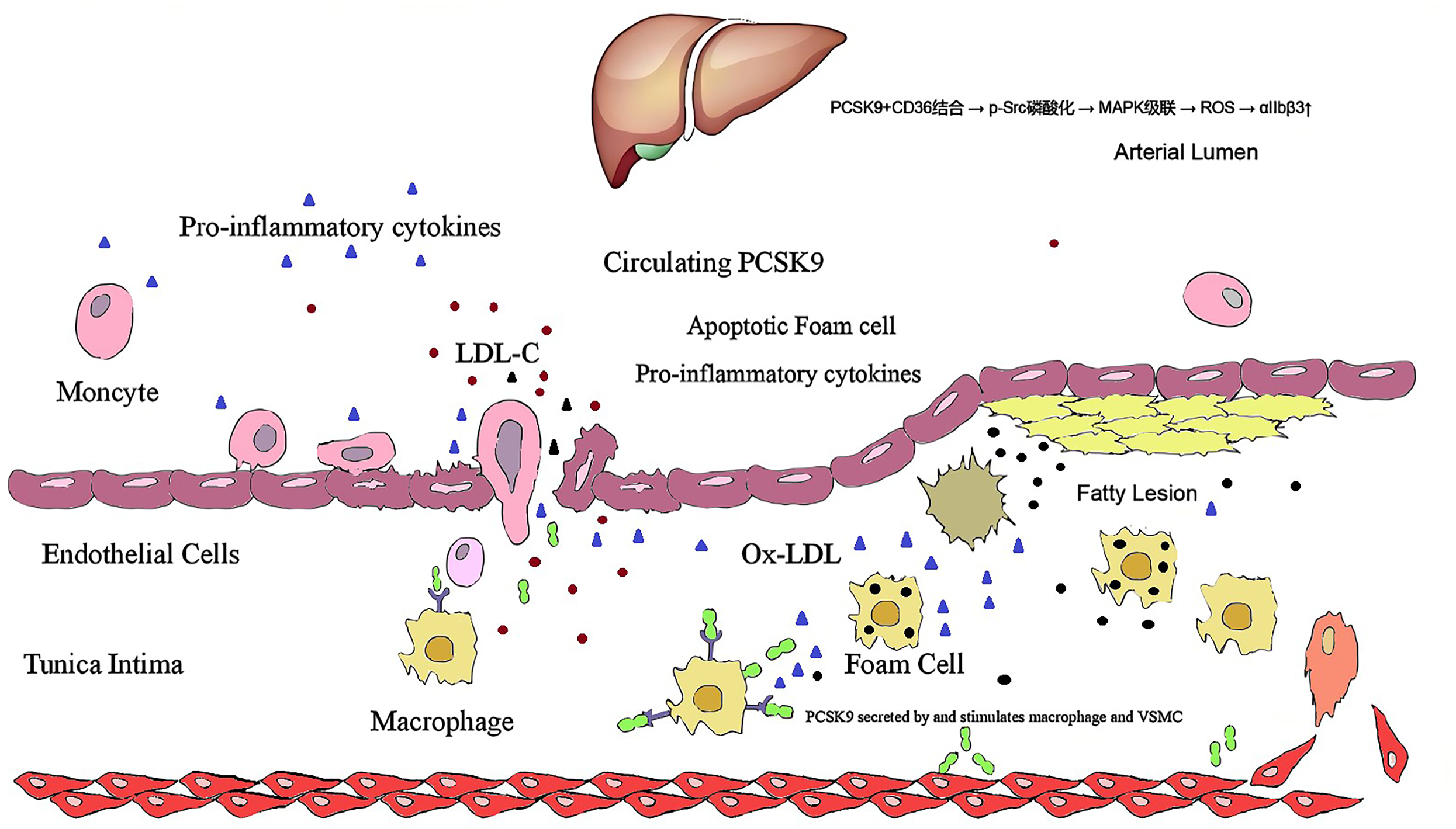
Foam cell formation. In vitro studies show PCSK9 enhances CD36-mediated LDL uptake while suppressing ABCA1-dependent cholesterol efflux in macrophages, promoting lipid accumulation. 35 However, these findings are derive from simplified cell culture systems lacking plaque microenvironmental complexity. Inflammatory amplification. PCSK9 induces the expression of IL-6, TNFα, VCAM-1, and ICAM-1 in macrophage cultures. 36 Clinically, plasma levels of these markers are elevated in PAD patients versus controls, though PCSK9's direct contribution remains correlative. Monocyte/macrophage recruitment. In ApoE−/− mice, overexpression of PCSK9 resulted in a 21% increase in intraplaque Ly6C −/− monocyte infiltration. 37 In Ldlr−/− venous grafts, AAV-PCSK9 exacerbated macrophage accumulation and MMP activity. 17
These murine models may not fully replicate human plaque biology due to species-specific differences in immune cell trafficking. Plaque instability implications. Foam cell accumulation drives lipid core expansion, which is a key feature of plaque vulnerability observed in human pathology. However, the direct role of PCSK9 in human plaque destabilization requires validation through clinical specimens.
PCSK9 regulated platelets to promote atherosclerosis development
Platelets critically sustain atherosclerosis progression through the inflammatory and thrombotic pathway. Clinical evidence suggests associations between PCSK9 and platelet hyperactivity. A prospective observational study (n = 178) showed a significant correlation between PCSK9 levels and platelet reactivity (r = 0.30; p = 0.004). Patients with higher PCSK9 exhibited markedly elevated platelet reactivity compared to those with lower levels (p = 0.02). 38 A cross-sectional study (n = 115) revealed positive correlations between serum PCSK9 and platelet activation markers, including P-selectin expression, GPIIb/IIIa activation, and light transmission aggregometry responses under various stimuli. 38 Plasma PCSK9 also correlated with platelet count (PLT; r = 0.218, p < 0.001) and plateletcrit (PCT; r = 0.250, p < 0.001). A prospective observational trial (n = 620) demonstrated that adding PCSK9 monoclonal antibodies to ticagrelor-based dual antiplatelet therapy significantly reduced platelet reactivity and improved cardiovascular outcomes. 39
PCSK9-mediated microcirculatory dysfunction
In vitro studies propose a mechanistic link between PCSK9 and platelet-driven microvascular impairment. (a) CD36-Src/MAPK-ROS axis. PCSK9 binding to platelet CD36 activates Src kinase and MAPK signaling in isolated human platelets, increasing ROS production and potentiating activation. 40 (b) Platelet hyperreactivity. In vitro studies confirmed that PCSK9 potentiated agonist-induced platelet aggregation, dense granule ATP release, integrin αIIbβ3 activation, α-granule secretion, platelet spreading, and clot retraction. 40 These findings derive from static platelet suspensions lacking endothelial/vascular wall interactions. (c) TLR4/NF-κB amplification. PCSK9 amplified ADP-induced aggregation via TLR4/NF-κB in platelet cultures. 41 Given species differences in TLR4 signaling, human in vivo relevance requires verification.
Therapeutic roles of PCSK9 inhibitors in PAD management
LDL-C reduction and cardiovascular protection
Large-scale randomized trials demonstrate cardiovascular benefits of PCSK9i in PAD subgroups. In a secondary analysis of the FOURIER trial, 3642 PAD patients were evaluated. The results revealed that evolocumab significantly reduced the risk of major adverse cardiovascular events (MACE), including cardiovascular death, myocardial infarction, stroke, hospitalization for unstable angina, or coronary revascularization. After 2.5 years of follow-up, the risk of the primary endpoint was reduced by 21% (HR 0.79, 95% CI 0.66–0.94). 42 Furthermore, a subgroup analysis of the ODYSSEY OUTCOMES trial 43 corroborated the cardiovascular benefits of alirocumab in PAD patients. It showed a marked reduction in cardiovascular event risk. These large-scale clinical trials suggested that PCSK9is may serve as an adjunctive therapy to statins to further lower LDL-C levels and mitigate cardiovascular risk in PAD patients.
Reduction of major adverse limb events
Beyond cardiovascular protection, landmark trials suggest limb-specific benefits of PCSK9is. In the PAD subgroup analysis of the FOURIER trial, evolocumab also significantly reduced the risk of major adverse limb events (MALE, defined as acute limb ischemia, major amputation, or urgent revascularization) by 42%. 42 This implied that PCSK9is may not only reduce systemic cardiovascular risk but also directly improve lower-extremity vascular function. However, in a separate study comparing alirocumab combined with statins versus statin monotherapy, although the alirocumab group achieved a significantly greater LDL-C reduction (mean difference [95% CI]: −49.8 [−66.1 to −33.6] vs. −7.7 [−19.7 to 4.3] mg/dL; p < 0.0001), no significant differences were observed in superficial femoral artery (SFA) plaque volume changes (+0.25 [−0.29 to 0.79] vs. −0.04 [−0.47 to 0.38] cm; p = 0.37), calf muscle perfusion (0.22 [were observed in superficial femoral artery (SFA) g; p = 0.46), or 6-minute walk distance (6MWD) after adjustment for baseline values. 44 These findings suggested that despite robust LDL-C lowering, the addition of alirocumab to statins did not significantly influence SFA plaque burden, limb perfusion, or functional capacity in PAD patients (Table 1).
Landmark studies of PCSK9 clinical trials in PAD.

Discussion
Although PCSK9i showed significant clinical benefits in PAD management, its precise positioning within therapeutic strategies remained under debate. Current guidelines recommend PCSK9i as a third-line therapeutic option for high-risk PAD patients who fail to achieve LDL-C targets despite high-intensity statin therapy combined with ezetimibe. However, emerging evidence of their pleiotropic effects (lipid-lowering, anti-inflammatory, plaque stabilization) and robust MALE reduction supports earlier deployment in progressive PAD, particularly with persistent hypercholesterolemia.
Therapeutic positioning of PCSK9 inhibitors in PAD: current controversies
Although PCSK9is demonstrate significant clinical benefits in PAD management, their optimal positioning within therapeutic algorithms remains debated. Current guidelines designate PCSK9i as third-line options for high-risk PAD patients failing to achieve LDL-C targets despite maximal statin-ezetimibe therapy. However, emerging evidence challenges this conservative approach. The FOURIER trial subgroup analysis revealed that evolocumab reduced MALE by 42% in PAD patients, with LDL-C reduction directly correlating with MALE risk reduction (HR 0.58 per 38.7 mg/dL LDL-C decrease; p < 0.001). This robust limb protection, coupled with pleiotropic effects (e.g. plaque stabilization via fibrotic cap thickening and lipid core reduction demonstrated in GLAGOV, anti-inflammatory actions via monocyte polarization), 45 supports earlier deployment in progressive PAD—particularly in cases of persistent hypercholesterolemia or high inflammatory burden.
Unresolved mechanistic and clinical knowledge gaps
PCSK9-Driven pathophysiology in PAD
The precise molecular mechanisms linking PCSK9 to PAD progression remain incompletely elucidated. Beyond canonical LDLR degradation, PCSK9 directly exacerbates vascular inflammation via TLR4/NF-κB activation, increasing Ly6C + monocyte infiltration and TNF-α/IL-6 secretion. 46 Notably, PCSK9 amplifies endothelial dysfunction by upregulating LOX-1, promoting ox-LDL uptake, and reducing nitric oxide bioavailability. 47 Crucially, PCSK9 independently associates with necrotic core progression in coronary plaques (p < 0.01 after adjusting for LDL-C), suggesting lipid-independent pathways relevant to PAD. However, its role in vascular calcification—a hallmark of PAD severity—requires deeper investigation. PCSK9 may mediate VSMC osteogenic switching via BMP-2/Runx2 activation, but human data are lacking.
Limitations of current clinical evidence
Long-term efficacy and safety data for PCSK9i in PAD are constrained by dependency on cardiovascular trial subgroups (e.g. FOURIER, ODYSSEY OUTCOMES). These studies exhibit three critical limitations. (a) Short follow-up durations (median 2.2 years), insufficient to capture long-term limb outcomes like amputation-free survival. 48 (b) Inadequate representation of advanced PAD phenotypes, such as CLI or diabetic microvascular disease. (c) Heterogeneity in background therapies, obscuring optimal PCSK9i dosing and combination strategies.
Imperatives for future research
The exact roles of PCSK9 in PAD pathogenesis remain incompletely elucidated. A deeper exploration of its molecular mechanisms was needed in disease progression. Long-term efficacy and safety data for PCSK9i in PAD patients remain limited, as current evidence primarily derives from subgroup analyses of cardiovascular outcome trials, with a paucity of large-scale, long-term follow-up studies specifically targeting PAD. Furthermore, optimal implementation strategies required systematic investigation, including treatment initiation timing, dose adjustment protocols, and combination therapies with other lipid-lowering agents, and so on.
Large-scale randomized controlled trials (RCTs) were urgently needed to evaluate the effects of PCSK9is on disease progression, limb-related events (e.g. CLI, amputation), and quality-of-life outcomes. Mechanistic studies should focus on clarifying PCSK9's involvement in PAD pathophysiology, particularly its interactions with vascular calcification and inflammatory cascades, which may unveil novel targets for early prevention and intervention. Additionally, research evaluating PCSK9 is combining synergistic strategies, such as antiplatelet regimens and supervised exercise rehabilitation, could optimize comprehensive disease management.
Conclusion
PCSK9 plays a multifaceted role in PAD pathogenesis, driven by genetic determinants, dyslipidemia, and direct vascular mechanisms. Gain-of-function variants elevate LDL-C and PAD risk, while loss-of-function mutations confer protection. Clinically, elevated plasma PCSK9 independently predicts PAD risk and correlates with disease severity. Beyond lipid regulation, PCSK9 drives endothelial dysfunction (via VEGFR2 degradation and impaired NO signaling), vascular inflammation (through LDLR-independent CAP1/Syk pathways), VSMC calcification, and platelet hyperreactivity. PCSK9is demonstrate significant cardiovascular and limb benefits in trials, partly via lipid-independent microcirculatory improvements. However, heterogeneity exists in local plaque/perfusion responses, underscoring unresolved pathophysiological nuances. Future research must clarify tissue-specific mechanisms and optimize targeted therapies for PAD progression.
Footnotes
Acknowledgments
The authors would like to thank Dr Zhu for their valuable guidance and suggestions. We are also grateful to all the participants for their technical assistance and helpful discussions.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Chongqing Sports Research Project (B202421); the National International Cooperation and Exchange Program of China (Grant No.81920108010).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
Data availability is not applicable to this article as no new data were created or analyzed in this study.