
Abstract
Objective
This study aimed to explore the genomic features of Halorubrum ezzemoulense strain TC23, an extreme halophilic archaeon isolated from the Ayvalık Saltern in Türkiye, with a focus on its halophilic adaptations and potential biotechnological applications.
Methods
Whole-genome sequencing was performed using the Illumina NovaSeq 6000 platform. In silico analyses were conducted using tools such as Prokka and eggNOG-mapper for structural and functional annotation, including gene ontology (GO), Clusters of Orthologous Groups (COG), and Kyoto Encyclopedia of Genes and Genomes pathway predictions. Taxonomic validation was performed with TYGS, and the genome was compared to related strains using digital DNA–DNA hybridization (dDDH), average nucleotide ıdentity, and phylogenomic tree construction.
Results
The genome comprised 3755 genes, including 3551 coding sequences. Key genes involved in osmoregulation, DNA repair, and salt tolerance were identified. Functional categorization based on COGs indicated pathways associated with hypersaline survival. Comparative genomic analysis confirmed the strain's placement within Hrr. ezzemoulense. Notably, the genome encodes extremozymes and stress-response genes relevant to high-salinity and oxidative environments.
Conclusions
The genomic insights provided by strain TC23 contribute to our understanding of halophilic adaptation mechanisms and highlight its potential in industrial applications requiring salt- and stress-resilient biocatalysts. This study supports future exploration of archaeal genomes as a resource for biotechnological innovation.
Introduction
Genomic analysis of halophilic microorganisms plays a key role in understanding how they adapt to extreme environments. These organisms, capable of thriving in high-salinity conditions, have evolved unique metabolic pathways and stress-response mechanisms over time. Exploring these adaptations at the molecular level not only enhances our understanding of basic biology but also opens the door to a range of biotechnological applications. 1 Genomic studies can uncover the genetic basis for the production of bioactive compounds, helping to connect adaptation strategies with real-world uses. Extracts derived from halophilic archaea show promising potential—as fluorescent agents, antioxidants, and natural colorants—which can be harnessed in the food and feed industries, as well as in cosmetics and healthcare.2–4
The genomes of halophilic microorganisms provide valuable insights into their ability to adapt to environmental and climatic changes. 5 Understanding the genetic adaptations of these organisms can provide valuable insights into strategies for future adaptation to environmental changes and contribute to sustainability efforts in areas such as agriculture, food security, and ecosystem health. 6 However, many of the genetic mechanisms underlying the biotechnological potential of halophilic microorganisms remain poorly understood. This highlights a significant gap in our current knowledge and limits the full exploitation of these organisms in industrial applications. In this study, we sequenced the whole genome of Halorubrum ezzemoulense strain TC23, which was isolated from the Ayvalık Salt Pan in Türkiye, and conducted a comprehensive in silico analysis. The primary goal was to uncover the genetic features that support the halophilic adaptations of this extremophilic archaeon and to explore its functional potential for industrial use based on genomic data. Through comparative analyses and functional classification, this work aims to provide new insights into the molecular mechanisms that enable Hrr. ezzemoulense TC23 to thrive in extreme environments.
Materials and methods
Isolation of halophilic archaeal strain
Brine samples were collected on 5 October 2021, from a solar saltern located in Ayvalık, Türkiye (GPS coordinates: 39.2593° N, 26.7194° E), under aseptic conditions. The sampling site is part of a traditional salt production facility, where high salinity creates favorable conditions for halophilic microbial diversity. Samples were transported to the laboratory within a few hours of collection, and 1 mL of the sample was directly inoculated into molten JCM 168 agar medium (50 mL) for the isolation of halophilic archaeal strains. The medium contained casamino acids (5 g/L), L-glutamic acid (1 g/L), yeast extract (5 g/L), trisodium citrate (3 g/L), MgSO₄·7H₂O (29.5 g/L), KCl (2 g/L), NaCl (150 g/L), FeCl₂·4H₂O (0.036 g/L), and MnCl₂·4H₂O (0.36 mg/L). The cultures were incubated at 39°C for 7 days. The selected temperature is within the optimal growth range (37–42°C) for Halorubrum species and was chosen to promote efficient colony development. 7 Pure colonies were selected from the plates and preserved in 15% glycerol at −20°C for further analysis.
Sample preparation for scanning electron microscopy
Fresh cultures of Hrr. ezzemoulense TC 23 were incubated at 37 °C for seven days. Cells were gently collected from the agar surface, spread onto coverslips, and left to air dry. They were then fixed overnight in 2% glutaraldehyde. After fixation, samples were air-dried and dehydrated through a graded acetone series (30%, 50%, 70%, and 90% for 10 min each, and 100% for 30 min) to remove residual moisture. Finally, the samples were coated with a thin Au-Pd layer using a Cressington Sputter Coater at 40 mA for 60 s and examined with a FEI Quanta FEG 250 scanning electron microscopy operating at 10 kV. Images were captured and processed using the xT Microscope Control software.
Genome sequencing, assembly, and annotation
Genomic DNA was extracted using the GeneMatrix Tissue and Bacterial DNA Kit following the manufacturer's instructions. DNA purity was assessed using a NanoPhotometer® spectrophotometer (IMPLEN, CA, USA), and concentration was determined with the Qubit® 2.0 Fluorometer (Life Technologies, CA, USA). DNA degradation and contamination were evaluated by 1% agarose gel electrophoresis. For library preparation, 1.0 µg of DNA was used, and sequencing libraries were constructed using the NEBNext® DNA Library Prep Kit for Illumina. DNA was fragmented to an average size of 350 bp, followed by end polishing, A-tailing, adapter ligation, and PCR enrichment. Library quality was verified using the Agilent 2100 Bioanalyzer, and quantified by real-time PCR. Paired-end sequencing (2 × 150 bp) was performed on the Illumina NovaSeq 6000 platform, achieving an average coverage of approximately 219-fold across the 3.73 Mbp genome.
Raw reads were quality-checked with FastQC (v0.11.9), and adapter sequences and low-quality bases were removed using Trimmomatic (v0.39). In this study, the newly sequenced strain TC23 was aligned against the reference genome of Halorubrum ezzemoulense strain Fb21 for comparative purposes using Bowtie2 (v2.4.4), and sorted with Samtools (v1.15). 8 Variant calling and filtering were performed with BCFtools, and functional annotation of variants was conducted using SnpEff (v5.0e). Consensus sequences were generated using vcf-consensus. Structural and functional annotation of the genome was performed with Prokka (v1.14.6) using the RefSeq database. Additionally, the genome was annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) for GenBank submission. For taxonomic classification, genome data were submitted to the Type (Strain) Genome Server (TYGS; https://tygs.dsmz.de), and classification was supported by the List of Prokaryotic names with Standing in Nomenclature (LPSN; https://lpsn.dsmz.de).9,10
In addition to phylogenetic analyses, genomic markers related to halophilic adaptation and biotechnological potential were also evaluated. In this context, genes related to osmoregulation (e.g. potassium transport systems), oxidative stress response (e.g. superoxide dismutase, catalase), carotenoid biosynthesis (crtI, crtY), and extremozymes were screened. In addition, the distribution of gene groups related to salt tolerance, stress adaptation, DNA repair, and transporter proteins was evaluated through functional classifications based on Clusters of Orthologous Group (COG) categories. These data provide insight into metabolic pathways and enzyme potentials of biotechnological interest.
Predicted proteins were annotated using eggNOG-mapper v2 with the eggNOG 5.0 database to assign GO terms, and Kyoto Encyclopedia of Genes and Genomes (KEGG) orthology. Annotation was performed with default parameters through the Galaxy Europe platform. 11 Functional enrichment analysis was based on GO biological process, molecular function, and cellular component terms, while KEGG pathway mapping identified major metabolic and biosynthetic routes.
Determination of closely related type strains
To identify the closest type strain genomes, two complementary methods were employed. Firstly, the MASH algorithm, a rapid approximation of intergenomic relatedness, was used to compare all genomes with all type strain genomes in the TYGS database. 12 Secondly, the 10 type strains with the smallest MASH distances were selected for each genome. Furthermore, an additional group of 11 type strains that are closely linked was identified using the 16S rRNA gene sequences. The user genomes were utilized to extract these sequences using RNAmmer. 13 Subsequently, each sequence was subjected to BLAST analysis against the 16S rRNA gene sequence of all the currently accessible 20957 type strains in the TYGS database. 14 The aforementioned method was employed as a substitute in order to choose the most suitable 50 type strains (based on the bitscore) for each genome of the user. Subsequently, accurate distances were calculated using the Genome BLAST Distance Phylogeny approach (GBDP) with the “coverage” algorithm and distance formula d5. 15 The distances were ultimately employed to ascertain the 10 most proximate type strain genomes for each genotype.
Pairwise comparison of genome sequences
The pairwise comparisons among the set of genomes were performed using GBDP for phylogenomic inference. Accurate intergenomic distances were inferred using the algorithm “trimming” using the distance formula d5 and reference. Each experiment consisted of 100 distance duplicates. The GGDC 4.0 was used to calculate digital DDH values and confidence intervals, using the suggested settings.10,15
Average nucleotide identity (ANI) analysis was performed to evaluate genomic similarity between strain TC23 and closely related Halorubrum genomes. FastANI v1.33 was used with default parameters (fragment length = 3000 bp; k-mer size = 16), using TC23 as the query genome and Halorubrum ezzemoulense strain Fb21 (GenBank accession: GCF_004126515.1) as the reference.
To explore the genomic diversity of Halorubrum ezzemoulense strain TC23, a pangenome analysis was performed using Roary (v3.13.0). 16 A total of 13 genomes from the genus Halorubrum (Hrr. ezzemoulense TC23, Hrr. ezzemoulense Fb21, Hrr. rubrum YC87, Hrr. lacusprofundi ATCC 49239, Hrr. kocurii JCM 14978, Hrr. distributum JCM 10247, Hrr. saccharovorum DSM 1137, Hrr. halophilum B8, Hrr. tropicale 5, Hrr. halodurans Cb34, Hrr. salipaludis WN019, Hrr. salinarum RHB-C, and Hrr. vacuolatum DSM 8800 were annotated using Prokka (v1.14.6) with the “kingdom Archaea” option prior to analysis. The annotated GFF3 files were used as input for Roary to identify orthologous gene clusters across genomes. Genes were categorized into core (present in ≥99% of genomes), soft core (95–98%), shell (15–94%), and cloud (<15%) gene groups. The resulting gene presence/absence matrix and summary statistics were further inspected to identify lineage-specific accessory genes.
Phylogenetic inference
Phylogenomic analysis was performed using GTDB-Tk v2.4.1 with the archaeal reference database release R226. The resulting Newick-formatted tree was visualized and annotated using the Interactive Tree Of Life v6 platform. Branch support values ranging from 0.8 to 1.0 were retained and displayed to highlight well-supported clades. 17
Results
Genome assembly and general features
The complete genome of Halorubrum ezzemoulense TC23 comprises 3,726,859 base pairs with a G + C content of 66.45%. A total of 3755 genes were annotated, including 3551 high-confidence coding sequences (CDSs), 81 RNA genes (2 5S, 2 16S, 2 23S rRNAs, 73 tRNAs, and 2 ncRNAs), and 123 pseudogenes. Detailed genome properties and the distribution of genes into functional COG categories are presented in Table 1. A complete list of annotated gene names, protein functions, sequence lengths, and COG classifications is provided in Supplementary Table 1.
Number of genes associated with general COG functional categories.

COG categories with zero hits were also assessed but are not shown in the table.
COG: Clusters of Orthologous Group.
The scanning electron microscope image of the species is given in Figure 1.

The scanning electron microscope (FEI Quanta FEG 250 SEM) image of Halorubrum ezzemoulense strain TC23 (Operating at 15 kV, 80.000X). SEM: scanning electron microscopy.
Structural and functional annotation
Genome annotation was performed using the NCBI PGAP 18 and Prokka. 19 The predicted protein-coding genes were classified into functional categories, including membrane transport, regulatory, and metabolic roles, alongside a subset of hypothetical proteins.
Comparative variant analysis between strain TC23 and the reference strain FB21 identified 14,123 high-confidence single-nucleotide polymorphisms (SNPs) and 365 insertions/deletions (INDELs). The transition/transversion (Ti/Tv) ratio of 3.19 suggests a mutational profile consistent with neutral evolution, indicating microdivergence without major structural rearrangement.
Taxonomic and phylogenomic analysis
Initial 16S rRNA sequencing revealed 95.69% identity to Hrr. ezzemoulense, suggesting potential novelty. However, genome-based analyses using TYGS indicated a digital DNA–DNA hybridization (dDDH) value of 88.7% to the type strain Hrr. ezzemoulense DSM 17463, confirming species-level identity. 20 Clustering based on a 70% dDDH threshold revealed that strain TC23 belongs to one of nine distinct species clusters and one of 10 subspecies clusters. FastANI analysis further supported this affiliation, showing 99.55% identity with strain Fb21.
The phylogenomic tree depicting the relationship of TC23 to other Halorubrum strains is shown in Figure 2. Species and subspecies cluster assignments are listed in Tables 2 and 3.

Whole-genome-based phylogenomic tree of Halorubrum strains, constructed using GTDB-Tk v2.4.1 with the archaeal reference database release R226. Branch support values between 0.8 and 1.0 are indicated by blue circles. Tree visualization and annotation were performed using iTOL v6. The genome of strain TC23 is highlighted and Haloarcula marismortui was used as the outgroup to root the tree. iTOL: Interactive Tree Of Life.
Genomic and taxonomic characteristics of related species and subspecies.
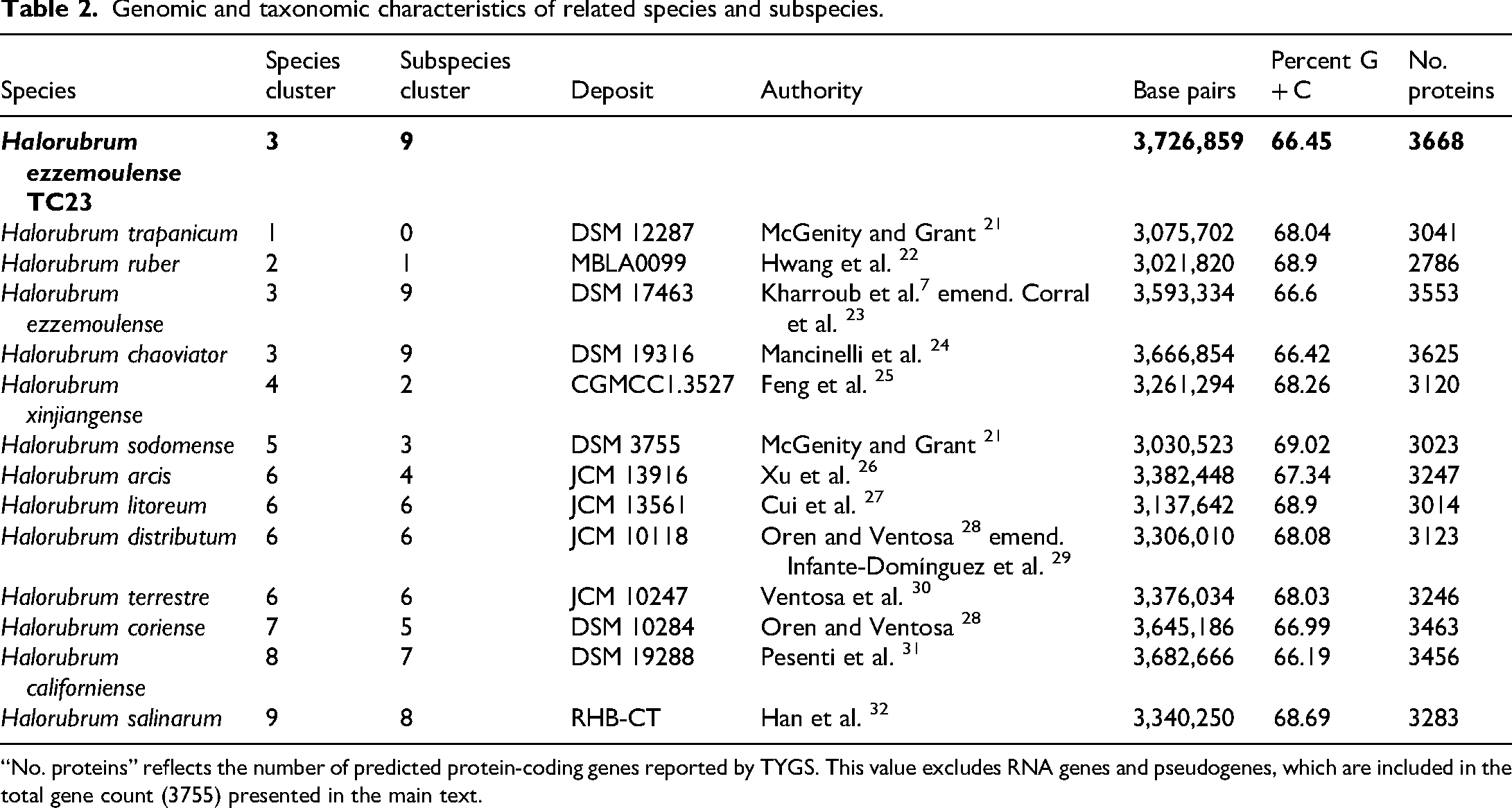
“No. proteins” reflects the number of predicted protein-coding genes reported by TYGS. This value excludes RNA genes and pseudogenes, which are included in the total gene count (3755) presented in the main text.
Pairwise comparisons of Hrr. ezzemoulense TC23 vs. type strain genomes.

Formula d4: sum of all identities found in high-scoring segment pairs (HSP) divided by overall HSP length.
dDDH: digital DNA–DNA hybridization; HSP: heat shock protein.
Evaluation of biotechnological potential via comparative genomics
A pangenome analysis of 13 Halorubrum strains revealed a total of 32,675 gene clusters. Of these, only 23 were core genes, and ∼80% were classified as cloud genes, highlighting the genus's open and dynamic pangenome structure. Although TC23 did not harbor any unique genes, a subset of 22 accessory genes were shared exclusively with strain Fb21. Notably, several of these genes were annotated as hypothetical proteins, while others were associated with membrane transport (ABC transporters), regulatory proteins (transcriptional regulators such as TrmB and Ptr2), or metabolic functions (enzymes involved in thiamine or NAD biosynthesis) (Supplementary File 1).
Discussion
The dDDH analysis revealed a high degree of genomic similarity between strain TC23 and Hrr. ezzemoulense DSM 17463, with a dDDH value of 88.7%. This finding affirms the strain's taxonomic classification within the Hrr. ezzemoulense species, despite initial 16S rRNA gene sequence analysis suggesting a potential new species. This result highlights the necessity of comprehensive genome sequencing over 16S rRNA-based analyses for accurate microbial classification.
Complete genome sequencing of Hrr. ezzemoulense strain TC23 provides valuable insights into the genetic basis for its extreme halophilism. This strain has a high G + C content of 66.45% that is typical among extremophiles; it facilitates DNA structure stabilization at high-salt conditions. 33 This genomic feature is crucial for maintaining cellular functions and integrity in environments where osmotic stress is prevalent.
A significant discovery in this study is the identification of 3551 coding sequences (CDS), including genes crucial for salt tolerance, osmoregulation, and DNA repair mechanisms. These genes play a vital role in the organism's ability to endure hypersaline conditions (Supplementary File 1). Although classical genes for glycine betaine and trehalose biosynthesis were not identified in our genomic analysis, the presence of multiple potassium transport-related genes suggests the existence of regulatory mechanisms involved in osmotic balance. The dataset provided highlights several genes that are crucial for the survival and adaptation of halophilic archaea in extreme environments. Among these, specific genes such as sensory rhodopsins play pivotal roles. Sensory rhodopsins, together with their associated transducer proteins (Htr2), are involved in light-dependent signal transduction that governs phototactic responses. 34 Rather than directional movement toward or away from light in a eukaryotic sense, these systems enable halophilic archaea to modulate their motility in response to changes in light intensity or quality. Heat shock proteins (HSPs) play a significant role in the stress response of halophilic archaea. 35 Among them, the small heat shock protein HSP16.5, identified in the TC23 genome (Supplementary File 1), is essential for protecting cells against environmental stresses such as high temperature and osmotic pressure. HSPs function by assisting in protein folding, preventing aggregation, and ensuring proper protein conformation under stress conditions. This capability is vital for maintaining cellular homeostasis and survival in fluctuating and extreme environments. In parallel, the presence of antioxidant enzymes such as superoxide dismutase and catalase further enhances the organism's resilience by mitigating oxidative damage.
One of the key genes identified is FilI (Methanogenesis Regulatory Histidine Kinase), which plays a pivotal role in the regulation of methanogenesis. This gene is crucial for the pathway that converts organic substrates into methane, a process vital for energy production in anaerobic conditions. The presence of the FilI gene may point to broader signaling or metabolic regulatory roles, although active methane production in Halorubrum remains unverified. The enrichment of pathways such as ABC transporters and two-component systems suggests that strain TC23 is well-equipped to sense and adapt to extreme osmotic stress, a hallmark of halophilic archaea. The abundance of genes involved in oxidative phosphorylation highlights its potential for efficient energy production in extreme environments.
The SNP analysis for Halorubrum ezzemoulense TC23 and FB21 strains showed substantial genetic variation that likely underpins differences in their environmental adaptations and metabolic functions. High-impact variants may be especially important for understanding critical functional divergences while moderate to low-impact variants contribute to wider range of genomic as well as phenotypic diversities.
KEGG pathway mapping assigned a substantial proportion of the predicted proteins to well-defined metabolic and biosynthetic pathways. The five most prominent pathways were “Metabolic pathways” (ko01100, 466 genes), “Biosynthesis of secondary metabolites” (ko01110, 227 genes), “Biosynthesis of antibiotics” (ko01130, 186 genes), “Microbial metabolism in diverse environments” (ko01120, 157 genes), and “Biosynthesis of amino acids” (ko01230, 107 genes) (Figure 3). These pathways are essential for maintaining cellular homeostasis under hypersaline conditions and point toward the biotechnological potential of strain TC23 in producing secondary metabolites and stress adaptation molecules.

Top 5 KEGG pathways identified in Halorubrum ezzemoulense strain TC23, illustrating predominant metabolic and biosynthetic capabilities. ko01100: Metabolic pathways. (ko01110: Biosynthesis of secondary metabolites, ko01130: Biosynthesis of antibiotics, ko01120: Microbial metabolism in diverse environments, ko01230: Biosynthesis of amino acids). KEGG: Kyoto Encyclopedia of Genes and Genomes.
A notable proportion of annotated genes lacked assignment to specific KEGG pathways. These genes may represent hypothetical proteins, novel enzymes, or yet-uncharacterized metabolic functions, underscoring the need for future functional validation and expansion of reference databases for halophilic archaea.
GO annotation provided further insight into the functional repertoire of strain TC23. The most enriched GO terms were related to catalytic activity (GO:0003824), metabolic processes (GO:0008152), and cellular processes (GO:0009987) (Figure 4). This distribution reflects a genome specialized in enzymatic and metabolic functions necessary for survival and growth under extreme environmental conditions.

Distribution of the top Gene Ontology (GO) terms assigned to Halorubrum ezzemoulense strain TC23, highlighting predominant molecular functions and biological processes. (GO:0008152: Metabolic process, GO:0009987: Cellular process, GO:0071704: Organic substance metabolic process, GO:0044238: Primary metabolic process, GO:0008150: Biological process, GO:0003824: Catalytic activity, GO:0003674: Molecular function, GO:0044237: Cellular metabolic process.).
Moreover based on the functional annotation provided by the NCBI PGAP, Halorubrum ezzemoulense TC23 was found to encode the enzyme porphobilinogen synthase (GO:0004655), which catalyzes the conversion of two molecules of δ-aminolevulinic acid into porphobilinogen—a critical precursor in the tetrapyrrole biosynthetic pathway (GO:0033014). This pathway leads to the production of essential cofactors such as heme, siroheme, and cobalamin (vitamin B12), which are vital for redox reactions, stress responses, and metal ion homeostasis. The presence of this enzyme suggests that strain TC23 may possess the metabolic capability to synthesize bioactive molecules with potential biotechnological applications, particularly in oxidative environments. Furthermore, several additional GO terms annotated through the NCBI pipeline—such as response to oxidative stress (GO:0006979), response to salt stress (GO:0009651), oxidoreductase activity (GO:0016491), and iron ion binding (GO:0005506)—underscore the strain's genomic adaptation to hypersaline and stressful conditions. These features not only reinforce the ecological robustness of Hrr. ezzemoulense TC23 but also highlight its promise as a source of stress-resilient biocatalysts and specialized metabolic pathways for industrial and environmental biotechnology.
The open nature of the Halorubrum pangenome observed in this study is consistent with previous reports on Hrr. ezzemoulense, where extensive genomic plasticity and clade-specific genomic islands were identified across 47 isolates. 36 Although no genes were found to be uniquely present in strain TC23, a distinct set of accessory genes was shared with Fb21 but remained largely absent from other Halorubrum genomes. These genes, some encoding hypothetical proteins and others involved in potential regulatory or membrane-associated functions, may represent strain-level adaptations that contribute to ecological differentiation.
The comprehensive genomic data generated in this study provide a valuable resource for future research aimed at exploring the biotechnological potential of Hrr. ezzemoulense. The unique enzymes and metabolic pathways identified in this extremophile can be harnessed for various industrial applications. For instance, enzymes with high salt tolerance and stability can be utilized in industrial processes requiring harsh conditions, such as high-salinity bioreactors.
Conclusion
This study presents a comprehensive genomic analysis of Halorubrum ezzemoulense strain TC23, highlighting its adaptation strategies to hypersaline environments. The complete genome revealed a compact core structure with substantial SNP-based microdivergence, suggesting evolutionary fine-tuning without large-scale gene gain or loss. Key genes associated with osmotic balance, oxidative stress response, and phototaxis were identified, reinforcing the organism's resilience under extreme conditions. Notably, the presence of carotenoid biosynthesis pathways, extremozymes, and stress-related regulatory systems points to the strain's potential in industrial applications that demand salt- and stress-tolerant biocatalysts. These functional capabilities, coupled with porphobilinogen synthase-related cofactor biosynthesis, further broaden the scope for biotechnological exploitation. The findings provide valuable insights into extremophile biology and lay the groundwork for experimental validation and future applied research. Overall, Hrr. ezzemoulense TC23 emerges as a promising candidate for harnessing extremophilic features in industrial biotechnology and environmental resilience strategies.
Supplemental Material
sj-docx-1-sci-10.1177_00368504251364316 - Supplemental material for Genomic insights into Halorubrum ezzemoulense strain TC23: Genetic basis for halophilic traits and biotechnological potential
Supplemental material, sj-docx-1-sci-10.1177_00368504251364316 for Genomic insights into Halorubrum ezzemoulense strain TC23: Genetic basis for halophilic traits and biotechnological potential by Fevziye Işıl Kesbiç in Science Progress
Footnotes
Acknowledgements
The author received no specific funding for this work. The study was conducted using the facilities of Kastamonu University.
Ethical statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Author’s contributions
FIK contributed to supervision, reviewing and editing, conceived and designed research, conducted experiments, analyzed data, and wrote the manuscript.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The complete genome sequences have been deposited in GenBank under accession numbers CP154831, CP154832, and CP154833 within BioSample number SAMN40624593 and BioProject accession number PRJNA1092433.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.