
Abstract
Objective
To develop a cost-effective and mechanically robust 3D collagen hydrogel system suitable for pressure-based culture, enabling physiologically relevant in vitro modeling of mechanical stress responses in cells.
Methods
A rat tail type I collagen-based hydrogel was formulated through optimized component ratios and cast into standard 24-well plates to form uniform gel columns. Endothelial cells was embedded and subjected to 30 mmHg pressure culture for up to 48 h. Gel morphology and fiber architecture were assessed via scanning electron microscopy. Cell viability, proliferation (Ki67 immunostaining), and tube formation ability were evaluated. A custom mechanical compression setup was used to apply and monitor sustained pressure.
Results
The hydrogel exhibited stable gelation, uniform porosity, and resistance to deformation under mechanical loading. SEM confirmed a consistent nanofiber network, with fiber diameter unaffected by 30 mmHg pressure. After 24-h pressure culture, the gel retained its height and structure. Endothelial cells remained viable but showed reduced proliferation and impaired tube formation under pressure, as indicated by Ki67 staining and angiogenesis assays.
Conclusions
This 3D collagen hydrogel provides a simple, cost-effective, and scalable alternative to complex bioprinting methods, supporting broader application of 3D cell culture in biomedical research.
Keywords
Introduction
Mechanical pressure fundamentally regulates cellular and tissue-level processes, with profound impacts on tumor progression, epigenetic modifications, embryonic development, and tissue remodeling.1–4 To investigate these mechanical effects, three-dimensional (3D) pressure culture gels have been developed to replicate in vivo mechanical microenvironments, serving as essential cellular models for applications in tissue engineering, regenerative medicine, and disease modeling. 5 In 3D cell cultures, cells exhibit diverse phenotypes, polarity, and asynchronous cell cycles, resulting in gene expression and metabolic profiles that more closely resemble the in vivo environment, whereas in 2D cultures, cells are confined to flat surfaces, leading to limited cell–cell interactions, synchronized growth and division, and a loss of structural complexity. 6 However, current 3D hydrogel fabrication methods are still limited by high cost, complex procedures, poor batch-to-batch consistency, limited bioactivity, and inadequate stability under long-term culture conditions.7,8
Currently, 3D in vitro models can be broadly categorized into scaffold-based and scaffold-free approaches. 9 Scaffold-based techniques generally involve the preparation of a physically stable biolink, followed by the use of 3D bioprinting to construct structured gels. 10 However, the need for stable gel formulations and expensive bioprinting equipment substantially increases production costs, while the multi-step preparation process contributes to technical complexity. 11
As a result, scaffold-free methods have also gained widespread use for constructing 3D cellular architectures. 6 These include suspending cells in hanging droplets, 12 utilizing ultra-low-adhesion surfaces to induce spheroid formation, 13 directly printing cell aggregates, 14 or employing magnetic levitation, in which cells are mixed with magnetic particles and aggregated under a magnetic field. 15 Nevertheless, each of these approaches presents limitations: hanging-drop and low-adhesion techniques often fail to generate mature tissue-like structures; direct bioprinting of cells or cell clusters is expensive, technically demanding, and difficult to scale up; and magnetic levitation poses potential cytotoxicity and limited cell compatibility due to the use of magnetic nanoparticles.
Collagen, a widely used biomaterial in 3D bioprinting, is available from various sources, each with distinct advantages and limitations. Animal-derived collagen (e.g. bovine, porcine, rat tail) offers strong bioactivity and supports cell adhesion and proliferation, but is limited by immunogenicity, batch variability. Recombinant human collagen provides improved consistency and low immunogenicity, but is expensive and technically demanding to produce. Marine-derived collagen is abundant and biocompatible, yet suffers from poor thermal and mechanical stability. Chemically modified collagens, such as methacrylated collagen, allow for enhanced printability and tunable properties, though chemical modifications may reduce bioactivity. 16 To simulate the mechanical compression conditions commonly used in hypertrophic scar treatment, we selected a culture pressure of 25 mmHg–30 mmHg, corresponding to the typical pressure exerted by commercial pressure garments.17,18 We employed the collagen scaffolds approach due to its advantage of providing a complex porous structure that closely resembles the native extracellular matrix (ECM). 19 In contrast to 3D bioprinting, which allows precise spatial patterning through layer-by-layer deposition, the collagen scaffold approach leverages the self-assembling fibrillogenesis of rat tail type I collagen under neutral pH, physiological temperature (37 °C), and appropriate ionic strength to form an interwoven 3D network. 20 This scaffold exhibits tunable stiffness and porosity, providing a supportive microenvironment for cell proliferation and differentiation. While it lacks the structural precision of bioprinting, its lower technical and equipment requirements make it a more accessible and scalable option for constructing physiologically relevant 3D culture systems. The key to successful implementation lies in optimizing the physicochemical conditions to ensure stable fiber formation and reproducibility.
By systematically optimizing the component ratios and concentrations, we successfully developed a novel 3D hydrogel system that supports cell proliferation, resists long-term immersion in culture media, and withstands mechanical compression-based culture. This hydrogel is not only cost-effective and easy to fabricate, but also possesses a complex porous architecture that enables long-term support of high-density cell cultures under immersed conditions. Furthermore, we present a comprehensive workflow that includes hydrogel preparation, enzymatic digestion for cell recovery, direct immunofluorescence staining of cells within the 3D matrix, and confocal microscopy imaging. This protocol underscores the potential of 3D culture systems to be widely adopted as more physiologically relevant alternatives to conventional 2D models in biomedical research. (Figure 1)

Preparation and workflow of the 3D rat tail type I collagen hydrogel system.
Materials and methods
Preparation of mouse tail type I collagen 3D culture gel
Cell preparation
Human microvascular endothelial cells (HMEC-1; Cat. No. ZQ0456) were purchased from Zhong Qiao Xin Zhou Biotechnology Co., Ltd (Shanghai, China). The identity of the cell line was confirmed by short tandem repeat (STR) profiling, which included the 8 core loci (D5S818, D13S317, D7S820, D16S539, vWA, TH01, TPOX, CSF1PO) and the amelogenin sex marker, among additional loci. The STR profile matched the expected reference for HMEC-1 (RRID: CVCL_0307), with no evidence of cross-contamination. Cells were confirmed to be mycoplasma-free prior to use. HMEC-1 cells were cultured in 10 cm dishes until reaching 85%-95% confluency (approximately 8 × 10^6 cells). Cells were trypsinized and resuspended in 6 ml complete culture medium. Each well for pressure culture was seeded with 1 ml cell suspension, yielding a final cell density of 1.3 × 10^6 cells/ml.
Preparation of 3D culture gel
In previous studies, the commonly used concentration of rat tail type I collagen was approximately 2 mg/ml, and required custom molds, increasing material requirements. 21 To simplify the procedure, we optimized the protocol by using standard 24-well plates as molds. At a final collagen concentration of 1.10 mg/ml, the system remained stable and consistently generated uniform gel structures. To minimize variability in hydrogel preparation, all formulations were prepared using reagents from the same lot number. Collagen solutions were pre-cooled on ice and mixed with other components in a consistent order under sterile conditions. Pipetting was performed using calibrated positive-displacement pipettes to ensure accurate volume transfer. Mixing time, temperature, and incubation duration were standardized across all replicates. The hydrogel mixtures were cast in standard 24-well plates with identical volumes (e.g. 300 μl per well) to ensure uniform gel column formation.
The gel components were added sequentially into each well of a 24-well plate: 42 μl of 10x PBS, 18 μl of 0.1 mol/L NaOH, 300 μl of mouse tail type I collagen (5 mg/ml, Hangzhou Xinyou Biotech Co., Ltd), and 1 ml of cell suspension in F12 DMEM containing 10% FBS. Critically, this order of addition must be maintained, as premature addition of NaOH to the acidic collagen solution results in uneven gel formation. The solution was mixed by pipetting up and down five times. The 24-well plate was incubated at 37°C with 5% CO2 for 10 min for gel formation. The resultant gel exhibited a semi-transparent appearance with a pale orange color and pH of 7.3–7.4.
Cell adaptation and gel transfer
The cells were allowed to adapt to the gel environment for 6–8 h before transfer to a Flexcell pressure culture plate. A modified 1 ml syringe tip, with the front 1 cm removed using a sterile scalpel, was used to transfer the gel to the pressure culture plate.
Pressure culture
Following gel transfer to the Flexcell pressure culture plate, 3 ml of serum-free medium was added to the outer chamber for pressure culture initiation. The gel maintained structural stability for 24 h during pressure culture and remained stable for up to 4 days.
Digestion of 3D hydrogels
Type I collagenase (Sigma) was dissolved in DMEM/F-12 to a final concentration of 1 mg/ml and sterilized through a 0.22 μm filter. The gels were transferred from the sponge ring to a 24-well plate and treated with 1 ml of collagenase solution at 37°C with gentle shaking for 20 min or until complete digestion. Cells were then collected by centrifugation at 1000 r/min for 3 min.
Scanning electron microscopy (SEM) analysis
Gel samples were longitudinally sectioned and fixed in 2.5% glutaraldehyde and 4% paraformaldehyde solution at 4°C for 3 h. After three 15-min buffer washes, samples were treated with 1% osmium tetroxide for 3 h, followed by three additional buffer washes. Samples underwent dehydration through a graded ethanol series (30–100%, 15 min each step), followed by three 15-min treatments with amyl acetate. Critical point drying was performed using carbon dioxide (35°C, 7.58 MPa). The dried samples were mounted on SEM stubs, gold-coated, and imaged.
Fiber diameter analysis was conducted using ImageJ software, incorporating Gaussian Blur filtering (sigma = 1 pixel), Rolling Ball background subtraction (radius = 600), and manual contrast adjustment for edge enhancement.
Immunofluorescence staining of cells in the gel
Gel columns were extracted using a 3 mm diameter puncher and flattened into thin films using absorbent paper. Rabbit anti-Ki67 antibody (1:100, Cat. No. 14-5698-82, RRID: AB_10854564; Thermo Fisher Scientific, Waltham, MA, USA) was used for immunofluorescence staining. The samples were incubated with antibodies in 96-well plates, followed by washing steps in 24-well plates with excess liquid removed using filter paper. After antifade reagent application, samples were transferred to confocal dishes for microscopic observation. Detailed antibody protocols were previously described. 18 Fluorescence images were acquired using a ZEISS LSM 880 with Airyscan super-resolution confocal microscope (Carl Zeiss Microscopy GmbH, Germany) from randomly selected fields. Image acquisition was performed using Zen 3.1 (Blue edition) software. Fluorescence intensity was quantified using ImageJ (NIH, USA), and data from three independent experiments were statistically analyzed using SPSS v28.0.1.1 (IBM, Armonk, NY, USA). Graphs were generated with GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA).
Angiogenesis assay
BD Matrigel was thawed at 4°C with pre-cooled pipette tips and Ibidi angiogenesis slides 24 h before use. Each well of the Ibidi slide's lower chamber was coated with 10 μl liquid Matrigel on ice and incubated at 37°C for 1 h in a humidified 10 cm culture dish. HDMECs retrieved from 3D culture hydrogel were resuspended in 1% FBS DMEM/F-12 (4 × 10^5 cells/ml), and 50 μl cell suspension was seeded per well. After 6 h of incubation at 37°C with 5% CO2, tube formation was documented using an Olympus automated upright fluorescence microscope.
Statistical analysis
Fiber diameter distributions were analyzed using Origin 2024 for histogram generation and normal distribution curve fitting, which indicated that the data were approximately normally distributed. All experiments were performed in three independent replicates. Mean fiber diameters among groups (0 mmHg 24 h, 30 mmHg 2 h, 30 mmHg 24 h) were compared using one-way ANOVA in IBM SPSS Statistics 28.0.1.1, followed by pairwise t-tests with Bonferroni correction. Statistical significance was defined as P < 0.05. The results are expressed as the means ± standard deviation (SD).
No formal sample size calculation was performed, as this study was designed as an exploratory in vitro experiment, in which three replicates per group are commonly accepted based on field standards and prior experience. 22
Results
Preparation and workflow of the 3D rat tail type I collagen hydrogel system
The gel preparation process in this experiment is detailed in Figure 1. The 24-well plates served as molds for pre-casting, resulting in gel columns with uniform volumes. These columns solidified within 15 min at 25 °C and maintained structural stability upon DMEM medium addition (Figure 2(A)). At a seeding density of 1.3 × 10^6 cells/ml, endothelial cells exhibited uniform distribution throughout the matrix after 6–8 h of incubation (Figure 2(B)).
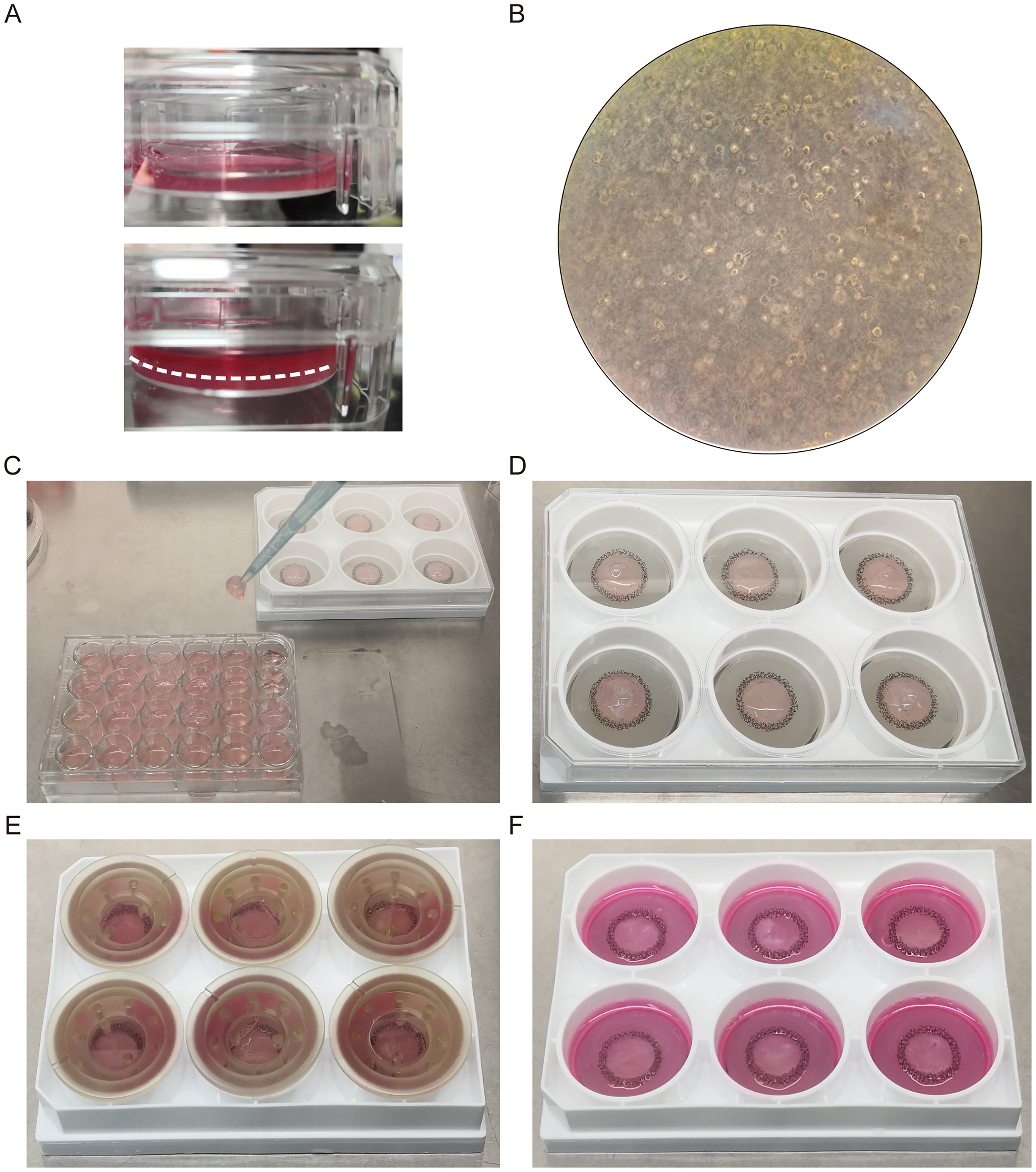
Structural stability and mechanical robustness of the collagen hydrogel system.
Structural stability and mechanical robustness of the collagen hydrogel system
The 3D hydrogel scaffold maintained structural integrity during mechanical manipulation, resisting suction force from a 1 ml pipette tip during plate transfers (Figure 2(C), (D)). After 24 h of mechanical pressure culture (30 mmHg, 37°C, 5% CO2), the hydrogel retained its original height of approximately 5 mm (Figure 2(E), (F)).
A custom-designed pressure culture system enabled precise control of mechanical loading on hydrogel scaffolds (Figure 3(A)). Fiber diameter analysis based on 50 randomly selected fibers from SEM images revealed distinct distributions under various pressure conditions (Figure 3(B) to (D)). At 0 mmHg for 24 h, fibers showed a mean diameter of 0.06454 ± 0.01342 μm with symmetric distribution. Under 30 mmHg for 2 h, the mean diameter increased to 0.06740 ± 0.02045 μm with right-skewed distribution. At 30 mmHg for 24 h, fibers exhibited a mean diameter of 0.06898 ± 0.01186 μm with enhanced uniformity.
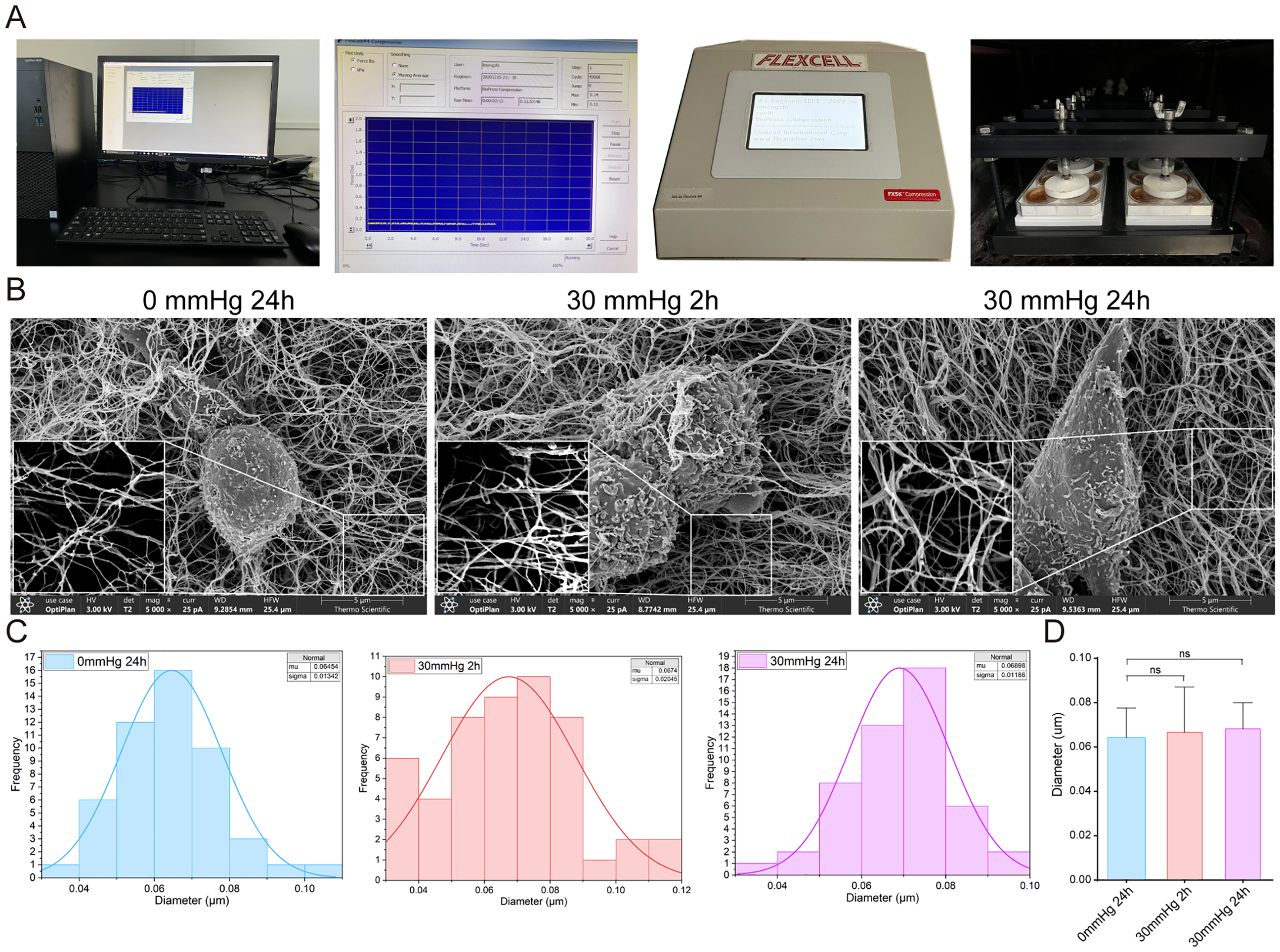
Mechanical performance and microstructural characterization of the hydrogel under sustained pressure.
Statistical analysis revealed no significant differences in fiber diameter across conditions. One-way ANOVA showed no significant variation among CTL, 2H, and 24H groups (F = 1.03, P = 0.361). Pairwise T-tests confirmed no significant differences between CTL vs. 2H (P = 0.411), CTL vs. 24H (P = 0.083), and 2H vs. 24H (P = 0.638).
The hydrogel scaffold maintained structural stability under applied mechanical pressure throughout the testing conditions. After 24 h of exposure to 30 mmHg pressure, the hydrogel exhibited consistent fiber distribution, stable structural height, and mechanical integrity without degradation, demonstrating its suitability for cell culture under mechanical stress conditions.
Biocompatibility and functional response of endothelial cells cultured in the 3D hydrogel
Endothelial cells were successfully retrieved from the hydrogel using type I collagenase digestion after culture with or without 30 mmHg mechanical pressure. The harvested cells formed tubes in angiogenesis 96-well plates, with reduced tube formation capacity observed in cells exposed to 30 mmHg pressure (Figure 4(A)).

Biocompatibility and functional response of endothelial cells cultured in the 3D hydrogel.
Immunofluorescence staining revealed clear cellular structures under confocal microscopy. Ki67 staining demonstrated significantly reduced proliferation in endothelial cells after 24 h of culture under 30 mmHg mechanical pressure compared to the control group without pressure (Figure 4(B)).
Discussion
3D cell culture models offer key advantages over conventional 2D systems by more closely mimicking the in vivo microenvironment.23,24 These models have been widely applied in tumor microenvironment, tumoroids, drug screening and toxicity testing, tissue engineering, stem cell differentiation.25,26 Current bioink systems used for 3D cell culture can be categorized into four types: natural polymer-based, synthetic polymer-based, hybrid, and functional bioinks.
Natural polymers such as collagen, gelatin, alginate, hyaluronic acid, and chitosan exhibit excellent biocompatibility and biodegradability, and can effectively mimic the native ECM to promote cell adhesion, proliferation, and differentiation. 27 However, they typically suffer from low mechanical strength, limiting their ability to maintain complex 3D structures. In contrast, synthetic polymers—such as polyethylene glycol (PEG), 28 polycaprolactone (PCL), 29 poly(lactic-co-glycolic acid) copolymer (PLGA), 30 and polyurethane (PU)—offer superior mechanical properties, structural stability, and tunable degradation rates. 31 Nonetheless, they often lack intrinsic bioactivity and require surface modifications to enhance cellular compatibility. Hybrid strategies aim to combine natural and synthetic polymers to leverage their respective strengths, improving overall performance. However, these approaches often involve complex formulation and crosslinking processes, typically requiring 3D bioprinting equipment for precise fabrication. Functional bioinks represent a more advanced category, incorporating responsive or bioactive components such as photoinitiators for in situ crosslinking, pH-responsive agents (e.g. polyacrylic acid) for simulating gastrointestinal or tumor environments, nano-hydroxyapatite (nHA) for mimicking bone ECM, or growth factors to induce angiogenesis. 32 While these enhancements improve the physiological relevance of 3D models, they also increase cost and technical complexity. Our study aims to develop a simplified, biocompatible hydrogel system that avoids specialized equipment and is suitable for large-scale applications.
We sought to develop a collagen-based hydrogel suitable for mechanical compression culture, with the initial goal of achieving a system that could support high cell density, tolerate prolonged immersion in culture medium, maintain structural integrity under mechanical stress, and allow efficient cell recovery. Through iterative optimization of formulation and pressure-based culture conditions, we established a hydrogel system with excellent structural stability, consistent batch reproducibility, and ease of preparation. Owing to its low cost, simplicity, and scalability, this system represents a promising alternative to more complex bioink-based platforms, offering a practical and accessible solution for broader 3D cell culture applications.
The Flexcell FX 5000 Compression System is widely used for applying mechanical forces to tissues and cells, yet few studies have provided detailed protocols for preparing 3D culture gels compatible with this system.18,33 Traditional methods, such as using a sponge ring in a pressure culture plate, often lead to liquid leakage and inconsistent gel volumes. To overcome these challenges, we developed a reliable 3D culture gel preparation method. Our approach introduces key improvements, including a pressure culture plate with a base area matching that of a standard 24-well cell culture plate. This ensures uniform 3D culture gels with consistent volume and cell density, ultimately enhancing experimental efficiency.
Our novel 3D culture gel, derived from rat tail type I collagen, offers several advantages, including ease of preparation, increased cell-loading capacity, superior mechanical integrity, and the ability to support cell proliferation and tubular structure formation. By refining the production of rat tail type I collagen, we have significantly reduced costs, making 3D culture more accessible and encouraging wider adoption of this preparation method. The gel remains stable in cell culture medium for extended periods, allowing for higher cell densities and improved viability—an essential factor in experiments involving sustained mechanical stress. Despite experiencing some compression after 24 h under mechanical pressure, the gel retained sufficient mechanical strength to maintain its integrity and height, demonstrating its suitability for mechanical loading applications. Scanning electron microscopy (SEM) revealed a uniform and stable nanofiber network, which is crucial for maintaining both mechanical stability and bioactivity. 34 Even after 24 h under 30 mmHg mechanical pressure, the nanostructure remained largely intact, highlighting the gel's durability and resistance to mechanical stress. In addition to experiments with endothelial cells, we also applied this gel system to human primary fibroblasts, demonstrating consistent gel formation and stability under the same pressure conditions. These results suggest the hydrogel may be compatible with multiple cell types, though further validation is needed. Mechanical cues in the cellular microenvironment play a key role in regulating cell behavior. In our study, Ki67 staining showed significantly reduced proliferation after 24 h of culture under 30 mmHg pressure compared to the non-pressurized condition, indicating that sustained mechanical stress can suppress cell proliferation. This study focused on short-term pressure exposure (up to 48 h), as longer durations at 30 mmHg markedly decreased cell viability.
This study has several limitations. Only one endothelial cell line (HMEC-1) was used for most experiments, and although preliminary tests were conducted with fibroblasts, additional cell types are needed to confirm broader applicability. Mechanical pressure was applied for a relatively short duration (up to 48 h), and long-term effects were not assessed due to reduced cell viability under sustained compression. Furthermore, no formal sample size calculation was performed, which may affect the statistical power of certain comparisons. Future studies with larger sample sizes, extended pressure durations, and power analyses will be essential to further validate and expand the system's applications.
We developed a simplified, cost-effective 3D collagen hydrogel system that supports high-density cell culture and maintains structural stability under mechanical compression. The hydrogel enables direct imaging and functional assays within a physiologically relevant 3D environment, offering a practical alternative to complex bioprinting-based models. This system holds promise for broader applications in mechanobiology, tissue engineering, and in vitro disease modeling.
Footnotes
Acknowledgements
This work was supported by the Shandong Provincial Natural Science Foundation (ZR2022MH290), the Shandong Provincial Natural Science Foundation Youth Program (ZR2023QH266), the Taian City Science and Technology Innovation Development Project (2020NS282).
Ethical considerations
Human microvascular endothelial cells (HMEC-1) were obtained from Zhong Qiao Xin Zhou Biotechnology (Shanghai, China). All experiments were conducted in accordance with institutional guidelines, and no ethical approval was required for the use of commercially available, anonymized cell lines.
Author contributions
Haiyan Wang and Mengzhi Li contributed to the study design and revised the manuscript. Bin Zhao, Zhengcheng Dang, and Lingling Li performed gel preparation, endothelial cell tube formation assays, scanning electron microscopy analysis, and immunofluorescence staining. Jing Gao was responsible for image processing and data analysis. Bin Zhao, Zhengcheng Dang, and Lingling Li contributed equally to this work as co-first authors and jointly participated in manuscript writing. All authors contributed to the article and approved the submitted version.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Shandong Provincial Natural Science Foundation Youth Program, Taian City Science and Technology Innovation Development Project, Natural Science Foundation of Shandong Province, (grant number ZR2023QH266, 2020NS282, ZR2022MH290).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All relevant data, including images, are included in this published article.