
Abstract
Objective
Epileptogenic tubers (ETs) contribute to the epileptogenesis in patients with tuberous sclerosis complex (TSC). High mobility group protein B-1 (HMGB1) triggers inflammatory responses that impact neuronal electrical activity. We aimed to study the role of HMGB1 in epileptogenicity in TSC-related epilepsy.
Methods
This study included both human and animal models. ETs were identified through comprehensive preoperative evaluations and postoperative seizure outcomes following tuberectomy. Immunofluorescence was performed to assess the positive area and fluorescence intensity of HMGB1 and IL-1β in ETs and non-ETs. TSC2-GFAP-CKO mice and epileptic models were established. Mice in the prevention group (postnatal day 14) received daily injections of monoclonal HMGB1 or IL-1β antibodies for 22 days. Mice in the intervention group (postnatal day 28) received identical treatments. Genetic and proteomic analyses of HMGB1 and IL-1β were conducted on both TSC patients and animal models.
Results
HMGB1 and IL-1β were significantly overexpressed in ETs compared to non-ETs. HMGB1 was predominantly nuclear in non-ETs but mainly cytoplasmic in ETs. In animal models, HMGB1 and IL-1β expression at both the gene and protein levels was significantly upregulated in the TSC2-deficient group compared to the wild-type group. IL-1β expression was lower in the HMGB1-intervention group than in the model control group. Seizure frequency was reduced in both HMGB1 and IL-1β prevention and intervention groups compared to the model control.
Conclusions
The interference of HMGB1 and IL-1β significantly reduced the frequency of seizure attacks in TSC. Epileptic seizures serve as key triggers for HMGB1 translocation and subsequent release into the cytoplasm, contributing to seizure recurrence.
Keywords
Introduction
Tuberous sclerosis complex (TSC) is a hereditary disorder with an autosomal dominant inheritance pattern, driven by pathogenetic variants in either the TSC1 or TSC2 gene. TSC can affect multiple organs, though childhood mortality often arises from neurological disorders, such as status epilepticus or sudden unexpected death in epilepsy. 1 Over 60% of individuals with TSC experience drug-resistant epilepsy with multiple types of seizures and associated intellectual disabilities, underscoring the clinical burden of refractory epilepsy in this population.2,3 Cortical tubers represent the hallmark brain lesions in patients with TSC-related epilepsy, which are categorized into epileptogenic tubers (ETs) and non-ETs. Resective surgery has proven effective in managing TSC-related intractable epilepsy, with 68% to 75% of patients achieving seizure freedom or significant seizure reduction (>90%) following the removal of ETs.4–6 Furthermore, 92.6% of patients undergoing resective epilepsy surgery have four or more cortical tubers in their brain; however, not all of these are classified as ETs. 6 The number and relative volume of cortical tubers stabilize after the first year of life, with no subsequent increases. Additionally, consistent locations of interictal epileptiform discharges on scalp electroencephalograph (EEG) further support the stability of ETs over time in patients with TSC-related epilepsy.7,8 There is no difference in the distribution of cortical tubers or ETs across various brain regions, and no significant genetic differences have been detected between ETs and non-ETs within the same patient. 3 These findings suggested that mechanisms beyond TSC-related mutations might contribute to the development of TSC-related epilepsy.
The mechanisms underlying epilepsy are complex, with a strong connection between the neuroimmune inflammation and the development of the disorder.9,10 High mobility group protein B-1 (HMGB1) is a nuclear protein that plays a critical role in regulating various DNA-related processes. Meanwhile, IL-1β plays a pivotal role in injury response and inflammation. 11 HMGB1 and IL-1β are known to influence the electrical activity of neurons and glial cells, further contributing to localized inflammatory responses.11–13 Seizures are frequently associated with elevated levels of IL-1β, emphasizing the impact of inflammatory cytokines on neuronal hyperexcitability and epileptogenesis.9,11 TLR4, a receptor involved in innate immunity, is expressed across various immune and non-immune cells in the mammalian body. 11 HMGB1 has been implicated in interacting with TLR4 receptors on the surface of neurons and glial cells in epilepsy.11–13 Epileptic seizures can initiate neuroinflammatory responses, exacerbating neuronal damage. This creates a vicious cycle in which inflammation, seizures, and brain injury perpetuate one another.14–17 The current prognostic methods of epilepsy recurrence and remission primarily rely on clinical assessments such as seizure semiology, magnetic resonance imaging (MRI), and electroencephalogram (EEG) recordings, however, the accuracy still requires improvement. 18 Therefore, inflammatory biomarkers offer a promising strategy to enhance diagnostic accuracy and deepen the understanding of epilepsy mechanisms linked to ETs. Notably, HMGB1 has emerged as a compelling candidate, showing significant potential for both epilepsy diagnosis and treatment.11,13,18 HMGB1 has gained attention as a major neuroinflammatory trigger, with evidence suggesting a regulatory axis involving HMGB1, TLR4, and IL-1β that may underlie seizure recurrence and resistance to treatment. 11
Despite the progress made in preclinical studies, much of the evidence linking HMGB1, TLR4, and IL-1β to epilepsy stems from animal models. Clinical validation in TSC-related epilepsy patients is needed to translate these findings into therapeutic strategies. This study investigates the relationship and underlying mechanisms of HMGB1, TLR4, and IL-1β axis in TSC-related epilepsy, with the goal of identifying novel targets for intervention and biomarkers in drug-resistant epilepsy.
Materials and methods
Preparation of patient's tissue
Cortical tuber specimens were obtained from 17 patients who underwent resective epilepsy surgery at Beijing Children's Hospital, comprising 17 pairs of ETs and non-ETs. The inclusion criteria for this study were as follows: (1) diagnosis of TSC based on clinical diagnostic criteria of TSC and confirmed TSC1 or TSC2 gene abnormality 19 ; (2) diagnosis of intractable epilepsy, defined by failure to respond to at least three different types of anti-seizure medications; (3) completion of preoperative evaluations, including stereo-EEG monitoring, conducted by a multidisciplinary team at our comprehensive epilepsy center in Beijing; (4) removal of the ET and adjacent non-ET during resective epilepsy surgery from September 2019 to July 2021. In all cases, tuberectomy (removal of the ET and nearby non-ET) was guided by intraoperative neuroimaging navigation. Patients were scheduled for follow-up one year post-surgery to evaluate seizure outcomes. An ET was defined as a cortical tuber confirmed as the seizure-onset zone through ictal stereo-EEG,6,20 while a non-ET referred to a neighboring cortical tuber without seizure activity on stereo-EEG monitoring. As there are currently no specific biological markers, imaging features, or pathological indications to definitively confirm whether the surgically resected tissue is an epileptogenic tuber, patients who continued to experience seizures after surgery were excluded to avoid misidentifying non-epileptogenic tubers as epileptogenic ones and subsequently using them for experimental research. This ensures that the cortical tissue collected postoperatively is indeed from the resected epileptogenic tuber. Patients experiencing seizures during the one-year follow-up were excluded from the study. Seizure outcomes were evaluated according to International League Against Epilepsy (ILAE) outcome classification: ILAE type 1 (seizure-free) and ILAE type 2–6 (not seizure-free). The extent of resection for both ETs and adjacent non-ETs was confirmed by comparing preoperative MRI T2-FLAIR images with postoperative MRI scans, as verified by a neurosurgeon and neuroradiologist. Tissue collection and manipulation were authorized by the Ethics Committee of Beijing Children's Hospital. Informed consent was collected from caregivers of all participants. The study was conducted in accordance with the Helsinki Declaration of 1975 as revised in 2024.
Animals and epilepsy model
TSC2 flox/flox-GFAP-Cre knockout (TSC2-GFAP-CKO) mice, with a C57BL/6 genetic background, had the TSC2 gene conditionally inactivated in glia driven by a glial fibrillary acidic protein (GFAP) promoter through the Cre-LoxP system. TSC2 flox/flox-GFAP-Cre knockout mice had a long history and were a typical model used to study TSC-related disease. No significant sex-based differences were observed, as previously described. 21 We used a total of 36 mice in this study, with no restriction on sex. The mice were aged between 6 and 8 weeks and weighed between 18 to 30 grams. Genotyping was conducted during the first week of life to confirm the presence of the targeted genetic modifications. The mice were housed in a controlled environment with a temperature of 22 ± 2°C, humidity of 40–60%, a 12-h light/12-h dark cycle, and provided with sufficient food and water to ensure their growth and health. The reporting of this study conforms to ARRIVE 2.0 guidelines. 22
The sample size (n = 6 per group) was determined using PASS software with the following parameters: Effect size (f) = 0.6, significance level (α) = 0.1 and power = 0.8. Additionally, we carefully considered ethical guidelines for animal experiments and resource feasibility. Antiepileptic drugs are only symptomatic treatments, which cannot prevent the occurrence or progression of epilepsy and do not correct the underlying brain abnormalities or mechanisms that lead to epilepsy. To develop more disease-targeted therapies, it may be necessary to target the underlying epileptogenic processes that occur during the latent period. Therefore, we have divided the experimental group into a prevention group and an intervention group. The epileptic model was established and randomly divided into six groups (n = 6 per group): wild-type control group, model control group (TSC2-GFAP-CKO mice), HMGB1 prevention group, IL-1β prevention group, HMGB1 intervention group, IL-1β intervention group (Figure 1A-1B). In prevention group, TSC2-CKO-Mice at postnatal day 14 (P14) were injected daily through the tail vein with either HMGB1 monoclonal antibody (8 μg/g of body weight) or IL-1β monoclonal antibody (11 μg/g of body weight) for 22 days.23–26 In intervention groups, TSC2-GFAP-CKO-mice at postnatal day 28 (P28) were injected through the tail vein with equivalent HMGB1 or IL-1β for a duration of 22 days. Epileptic mouse model was induced with pentylenetetrazole (PTZ) at a dose of 35 mg/kg via intraperitoneal injection for 22 consecutive days at P27. 27 Mice that exhibited Racine stage IV-V were considered valid model mice. 28 Additionally, we recorded latency to seizure onset to assess baseline seizure susceptibility, finding no significant differences among groups (P > 0.05). The seizure frequencies were recorded on postnatal day 50 (P50) and 57 (P57).
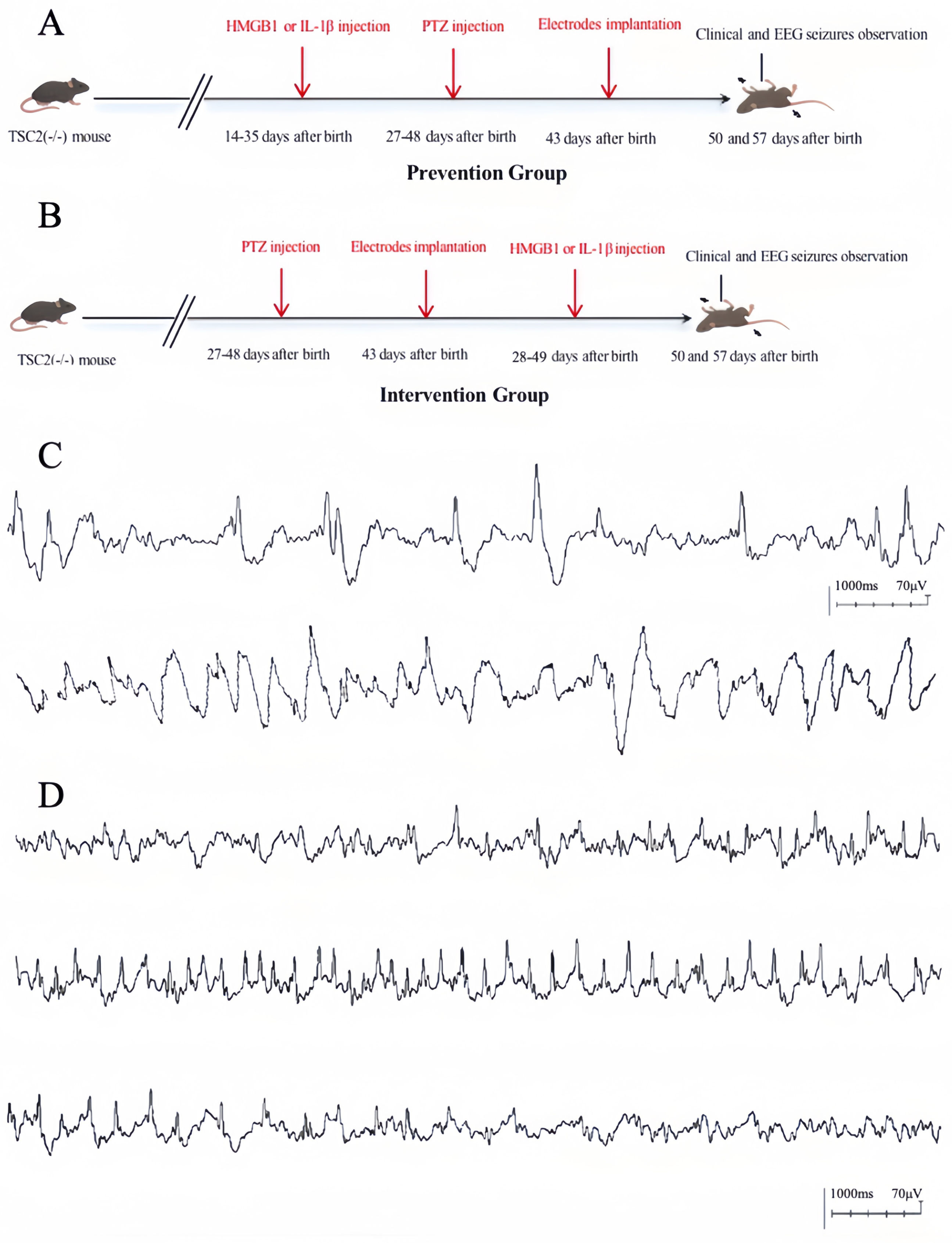
The interference flowchart of mice model in prevention and intervention group, the interictal epileptiform discharge and ictal EEG pattern. Figure 1A: Schematics depicting the experimental setup for model mice in the prevention group. Figure 1B: Schematics depicting the experimental setup for model mice in the intervention group. Figure 1C: The interictal epileptiform discharge on EEG was defined as abnormal discharge on EEG. Figure 1D: The ictal EEG was defined as continuous synchronous explosive electrical activity exceeding 2 Hz for more than 3 s.
Surgical procedure and EEG monitoring: Anesthesia was induced through intraperitoneal injection of isopentobarbital at a dose of 50 mg/kg body weight. Electrode implantation was performed on postnatal day 43 (P43). After securing the mouse on a surgical platform, the head skin was disinfected, and the skull was exposed. A 0.5 mm screw was inserted 2 mm anterior and 2 mm lateral to the anterior fontanelle to act as both the reference electrode and ground wire. Similarly, a 0.8 mm screw was placed 2 mm posterior and 2 mm lateral to the anterior fontanelle, reaching the cortex, and served as the recording electrode. Post-surgical care included daily injections of 2000 IU penicillin on the first three days following the procedure to prevent infection. Seven days after implantation, the mice were secured in a customized plastic fixator, and EEG recordings began with the mice in a fully awake state. To avoid time-related variability, EEG sessions were conducted between 9:00 and 11:00 AM at seven-day intervals. We recorded a baseline EEG during a non-seizure, awake state in the mice. This baseline data helps to define the normal brain activity patterns in the mice and serves as a reference for interpreting subsequent EEG data. The recording of epileptic discharges was defined as abnormal discharge on EEG, and the recording of continuous synchronous explosive electrical activity exceeding 2 Hz for more than 3 s on EEG was defined as seizure event (Figure 1C-1D). 29 By comparing the recorded EEG during seizure periods with the baseline, we are able to determine whether seizure discharges or other abnormal electrical activities have occurred.
After the experiment, brain tissues from all the mice were collected and processed according to their respective groups. Given the differences in clinical phenotypes between the TSC mouse model and humans, cortical tubers are rarely observed in mouse brains. 30 Wild-type and model (TSC2-GFAP-CKO) mice cortex were detected, rather than focusing on cortical tubers. The animals were euthanized using CO2, and death was confirmed by the absence of heartbeat and respiratory movements. We minimized the number of animals used through sample size calculations and adherence to the 3Rs, while ensuring their well-being by implementing humane endpoints, appropriate anesthesia and analgesia protocols, and following the Guide for the Care and Use of Laboratory Animals, 8th Edition. 31
Real-time polymerase chain reaction (Rt-PCR)
The primer sequences are presented in Supplementary Table 1. Following the thawing process, the tuber tissues were ground into powder, and total RNA was isolated using the TRIZOL RNA extraction kit (Tiangen Biotech, Catalog No. DP405–02). RNA concentration and purity were assessed through ultraviolet absorption with a NanoDrop® ND-2000, with DEPC water used for baseline calibration. Purity and RNA integrity were further verified by denaturing agarose gel electrophoresis. cDNA synthesis was carried out using the PrimeScript™ RT reagent kit with gDNA eraser (Takara Bio, Catalog No. RR047B). Real-time PCR was performed to quantify the target genes and internal reference for each sample, with triplicates used for each measurement by Applied Biosystems QuantStudio 6 system. The gene expression level was normalized to GAPDH expression, and calculated using the 2^-ΔΔCT method. Three independent biological replicates were used for the experiments.
Enzyme-linked immunosorbent assay (ELISA)
The levels of HMGB1, TLR4, IL-1β, and IL-1R in the tissue supernatant were measured using ELISA kits following the manufacturer's protocols (Thermo Fisher Scientific, USA). Absorbance at 450 nm was recorded for each well using a spectrophotometer, and protein concentrations were determined based on a standard curve. The final concentrations were obtained by adjusting for the dilution factor. Three independent biological replicates were used for the experiments.
Immunofluorescence
Immunofluorescence experiments are primarily used to detect and localize the expression and distribution of specific proteins in tissues. In this study, the main goal of the immunofluorescence experiments is to evaluate the expression of HMGB1, TLR4, IL-1β, and IL-1R during the process of epilepsy and their relationship with neuroinflammatory responses. Paraffin sections were dewaxed in water and subjected to antigen retrieval in a microwave oven using EDTA antigen retrieval buffer (pH 8.0). Once slightly dried, a histochemical pen was used to draw a boundary around the tissue. A spontaneous fluorescence-quenching agent was applied within the marked area for 5 min, followed by a 10-min rinse under running water. Bovine serum albumin was then added to the same area, and the sections were incubated for 30 min. The primary antibodies HMGB1 (1:250; Abcam; cat. no. ab79823), TLR4 (1:100; Abcam; cat. no. ab22048), IL-1β (1:50; Abcam; cat. no. ab315084), and IL-1R (1:50; Santa Cruz Biotechnology; cat. no. sc-66054) were incubated overnight at 4°C. The next day, after being washed with phosphate-buffered saline (PBS), sections were incubated with Alexa Fluor 568 and Alexa Fluor 488 (anti-rabbit IgG or anti-mouse IgG; 1:500) for 2 h at room temperature. The nuclei were counterstained with DAPI solution. Finally, the slides were sealed and observed under a fluorescence microscope for imaging. The sections were imaged at ×4 and ×40 using a Leica TCS SP8 confocal microscope. The pictures were captured by Leica LAS X. The area and fluorescence intensity of positive cells were automatically analyzed using Image-Pro Plus 6.0 software. HMGB1: Threshold set to 32–255, TLR4: Threshold set to 18–255, IL-1β: Threshold set to 23–255, IL-1R: Threshold set to 34–255.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism (version 10.2.0; GraphPad Software, Inc., San Diego, CA, USA). Results are presented as the mean ± SD. The tests employed include the t-test, Wilcoxon Signed-Rank Test, one-way ANOVA test, and Mann-Whitney U-test. Variance homogeneity was assessed with the f-test and Bartlett's test, while the Shapiro-Wilk test checked for normality. Tukey's correction was applied for multiple comparisons in the statistical tests. Nonparametric tests should be used when the data are not normally distributed or when variance is unequal. Statistical significance was defined as a two-tailed P-value < 0.05. To prevent bias, the authors remained blinded to the experimental protocol during both the experiments and statistical analysis.
Results
HMGB1 and TLR4 expression was increased in patients with TSC-related epilepsy and TSC2-GFAP-CKO mice
The relative RNA expression levels of HMGB1 (1.40 ± 0.32, P < 0.0001) and TLR4 (1.59 ± 1.01, P = 0.045) were measured in epileptogenic tubers (ETs) from patient samples. Both HMGB1 and TLR4 exhibited significantly higher expression in ETs compared to non-ETs (Figure 2A). To assess protein expression, ELISA was performed, revealing that HMGB1 (1083.13 ± 241.53) and TLR4 (3936.67 ± 1216.50) levels in non-ETs were significantly lower than those in ETs (HMGB1: 1340.62 ± 196.08, P = 0.017; TLR4: 5391.59 ± 872.02, P = 0.007) (Figure 2B). The differential expression of HMGB1 and TLR4 in ETs and non-ETs may play a role in the epileptogenesis of TSC patients.

The relative RNA and protein expression of HMGB1, TLR4, IL-1β, IL-1R in epileptogenic tubers (ETs) and non-epileptogenic tubers (non-ETs) (RNA :n = 17; protein: n = 10), and wild-type and control model mice group (n = 6). Figure A showed the relative RNA level of HMGB-1 (1.40 ± 0.32, t(17) = 5.574, P < 0.0001), TLR4 (P = 0.045), IL-1β (P = 0.005), and IL-1R (P = 0.009) were significantly lower in non-ETs compared to ETs. Figure B showed the protein level of HMGB-1 (t(18) = 2.617, P = 0.017), TLR4 (t(18) = 3.074, P = 0.007), IL-1β (t(18) = 2.140, P = 0.046), and IL-1R (P = 0.043) were significantly lower in non-ETs than ETs. Figure C showed the relative RNA level of HMGB-1 (P = 0.004), TLR4 (t(10) = 4.179, P = 0.002), IL-1β (t(10) = 6.411, P < 0.0001), and IL-1R (P = 0.004) were significantly lower in wild-type control group compared to model control group. Figure D showed the protein level of HMGB-1 (t(10) = 4.461, P = 0.001), TLR4 (t(10) = 6.558, P < 0.0001), IL-1β (t(10) = 3.449, P = 0.006), and IL-1R (t(10) = 6.108, P = 0.0001) were significantly lower in wild-type control group compared to model control group (*: p < 0.05; **: p < 0.01; ***: p < 0.001; ****p < 0.0001).
In the animal model, the relative RNA level of HMGB1 and TLR4 in wild-type control group was markedly lower compared to the model control group (HMGB1: 0.81 ± 0.50 vs 3.03 ± 1.42, P = 0.004; TLR4: 0.94 ± 0.66 vs 2.97 ± 0.99, P = 0.002) (Figure 2C). Similarly, proteomic analysis showed significantly lower protein levels of HMGB1 and TLR4 in the wild-type group compared to the model group (HMGB1: 997.24 ± 176.69 vs. 1438.87 ± 166.11, P = 0.001; TLR4: 2878.45 ± 317.21 vs. 4550.19 ± 537.83, P < 0.0001) (Figure 2D). Both HMGB1 and TLR4 were overexpressed in TSC2-deficient epileptic mice. This differential expression pattern may contribute to the development of epilepsy in TSC models.
IL-1β and Il-1R expression was increased in patients with TSC-related epilepsy and TSC2-GFAP-CKO mice
In patients with TSC, the average relative RNA expression levels of IL-1β (2.37 ± 2.02, P = 0.005) and IL-1R (2.43 ± 2.51, P = 0.009) were significantly higher in epileptogenic tubers (ETs) compared to non-ETs (Figure 2A). The protein content of IL-1β also showed a significant difference between ETs (253.16 ± 41.37) and non-ETs (196.16 ± 51.93) (P = 0.046). Similarly, the protein level of IL-1R was higher in ETs (3477.25 ± 741.68) than in non-ETs (2846.46 ± 805.50) (P = 0.043) (Figure 2B). These findings highlight the important role of inflammatory factors in the mechanism of epilepsy.
In the epileptic animal model, the relative RNA expression of both IL-1β and IL-1R was significantly lower in the wild-type control group compared to the model control group (IL-1β: 0.86 ± 0.23 vs. 2.53 ± 0.60, P < 0.0001; IL-1R: 1.07 ± 0.63 vs. 2.89 ± 0.96, P = 0.004) (Figure 2C). Likewise, protein expression levels of IL-1β and IL-1R were markedly reduced in the wild-type group compared to the model group (IL-1β: 171.30 ± 55.49 vs. 280.47 ± 54.17, P = 0.006; IL-1R: 1622.42 ± 212.63 vs. 2949.45 ± 487.88, P = 0.0001) (Figure 2D). These results indicate that IL-1β and IL-1R are significantly upregulated in TSC2-deficient epileptic mice, showing a distinct expression pattern compared to wild-type controls, which further supports their involvement in epilepsy pathogenesis.
The immunofluorescence expression pattern of HMGB1/TLR4 and Il-1R/Il-1β in patients with TSC-related epilepsy
Immunofluorescence staining was conducted to explore the microscopic expression pattern of HMGB1/TLR4 and IL-1R/IL-1β in ETs and non-ETs. The percentages of positive cells (positive area) expressing HMGB1, TLR4, and IL-1R were lower in the non-ETs compared to ETs (Figure 3). Besides, the fluorescence intensity of HMGB1, TLR4, IL-1β, and IL-1R also was lower in the non-ETs compared to ETs (Figure 3). Moreover, HMGB1 exhibited distinct localization patterns: it was primarily confined to the nucleus of neurons in non-ETs, whereas in ETs, it was predominantly expressed in the cytoplasm (Figure 3). These findings highlight the differences in the microscopic distribution and epileptogenic potential between ETs and non-ETs, suggesting a role for these distinct patterns in the pathogenesis of TSC-related epilepsy. The expression of HMGB1 and IL-1β may contribute to the heightened epileptogenicity observed in TSC-related epilepsy.
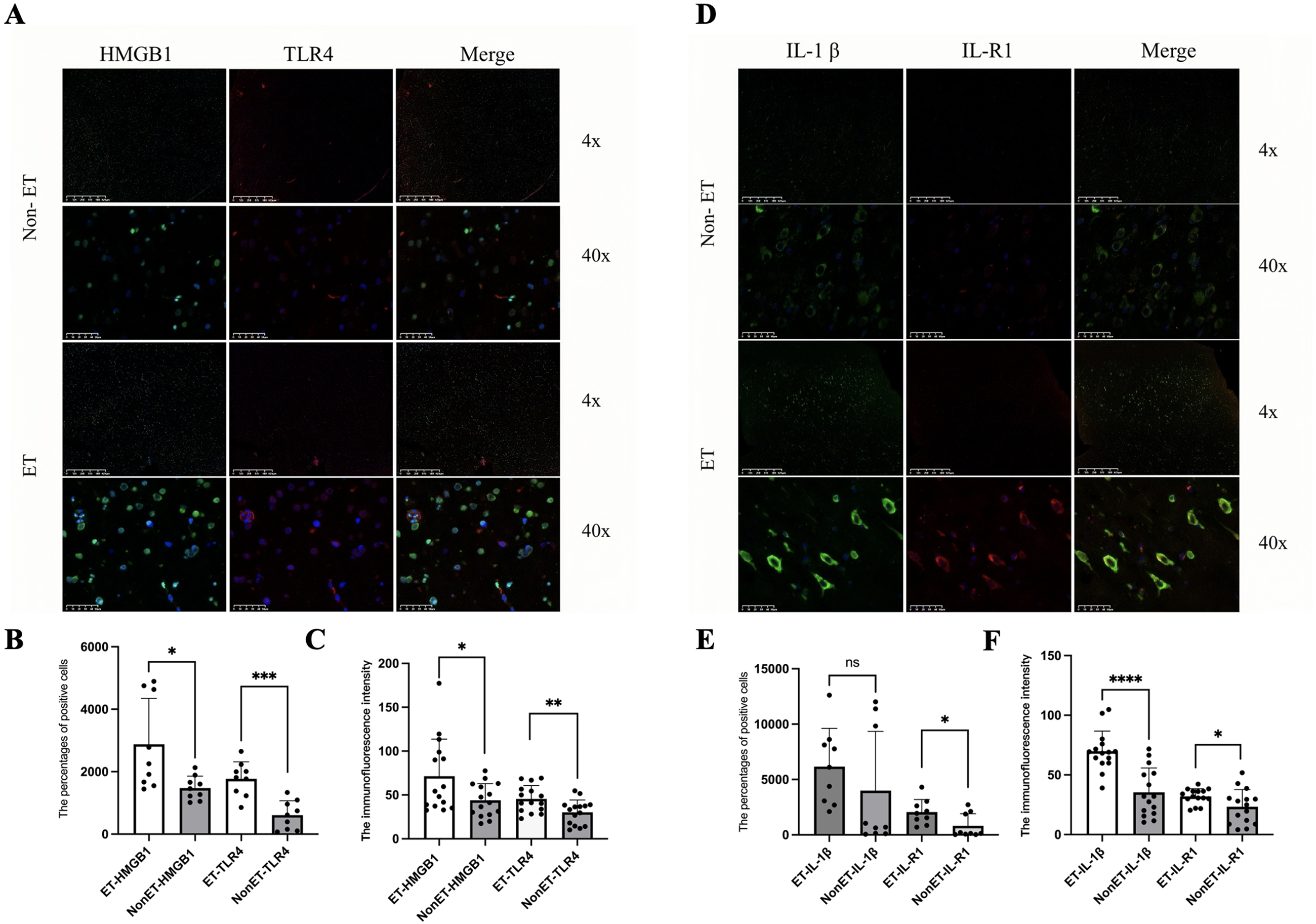
Immunofluorescence of HMGB1,TLR4, lL-1β and IL-1R in ETs and non-ETs. Positive expression of HMGB1 is green, and positive expression of TLR4 is red on the image of immunofluorescence in ETs and non-ETs (Figure 3A, magnifications: × 4 and ×40). The percentages of positive cells (positive area) (n = 9) of HMGB1 (t(16) = 2.770, P = 0.014) and TLR4 (t(16) = 4.884, P = 0.0002)were larger in ETs than them in non-ETs (Figure 3B) and, the fluorescence intensity (n = 15) of either HMGB1 (t(28) = 2.285, P = 0.030) or TLR4 (t(28) = 2.857, P = 0.008) was higher in ETs than them in non-ETs (Figure 3C). Positive expression of IL-1β is green, and positive expression of IL-1R is red on the image of immunofluorescence in ETs and non-ETs (Figure 3D, magnifications: × 4 and ×40). The positive area (n = 9) of IL-1R (P = 0.031) was larger in ETs than non-ETs (Figure 3E), and the fluorescence intensity (n = 15) of either IL-1β (t(28) = 5.055, P < 0.0001) or IL-1R (t(28) = 2.111, P = 0.043) were higher in ETs non-ETs (Figure 3F).
HMGB1 regulated inflammatory response via Il-1β in animal model
To further prove the preventive and therapeutic function of HMGB1 in epilepsy, TSC2-GFAP-CKO mice in both the prevention and intervention groups were administered monoclonal antibodies targeting HMGB1 or IL-1β via tail vein injection. Prophylactic interference with HMGB1 resulted in a significant suppression of IL-1β expression whereas, no noticeable reduction in IL-1β levels was observed in the HMGB1-intervention group (Figure 4). The relative RNA and protein levels of IL-1β were significantly decreased in IL-1β-prevention and intervention group compared to the model control group. Additionally, HMGB1 expression displayed a downward trend following interference with IL-1β. Overall, it was suggested that there may be an inflammatory signaling pathway mediated by HMGB1 targeting to IL-1β.

The expression pattern of relative RNA and protein of HMGB1, TLR4, IL-1β, IL-1R, and the frequency of abnormal discharge and seizure attacks in mouse model. (red line: P50; blue line: P57). Figure (5A-5D) shows the relative RNA content of HMGB-1, TLR4, IL-1β and IL-1R in wild-type control group, model control group, HMGB1 prevention group, IL-1β prevention group, HMGB1 intervention group, and IL-1β intervention group. The relative RNA content of IL-1β in HMGB1-intervention group was lower compared to model control group (t(10) = 2.645, P = 0.025) and higher than wild-type group. Figure (5E-5H) showed the protein levels of HMGB-1, TLR4, IL-1β and IL-1R in wild-type control group, model control group, HMGB1 prevention group, IL-1β prevention group, HMGB1 intervention group, and IL-1β intervention group. The protein content of HMGB1 (t(10) = 4.461, P = 0.001), TLR4 (t(10) = 6.558, P < 0.0001), IL-1β (t(10) = 3.449, P < 0.006), and IL-1R (t(10) = 6.108, P = 0.001) in wild-type control group was significantly lower than model control group. Figure (5I-5J): The frequency of seizure attacks at P50 in the HMGB1- prevention (P = 0.007) and -intervention (P = 0.027) groups was significantly decreased compared with the model control group. The frequency of seizure attacks at P50 in the IL-1β- prevention (P = 0.009) and -intervention (P = 0.046) groups were significantly decreased compared with the model control group. The frequency of abnormal discharge on EEG at P50 in the HMGB1- prevention (P = 0.007) and -intervention (P = 0.023) groups were significantly decreased compared with the model control group. The frequency of abnormal discharge on EEG at P50 in the IL-1β- prevention (P = 0.002) and -intervention (P = 0.011) groups were significantly decreased compared with the model control group. The frequency of abnormal discharge on EEG (P < 0.0001) and seizure attacks (P = 0.0004) at P50 in the wild-type control group were significantly decreased compared with the model control group. The frequency of abnormal discharge on EEG (P = 0.018) and seizure attacks (P = 0.021) at P57 in the wild-type control group were significantly decreased compared with the model control group (*: p < 0.05; **: p < 0.01; ***: p < 0.001; ****p < 0.0001).
The frequency of seizure attacks and abnormal discharge on EEG was decreased in prevention and intervention group in epilepsy animal model
In our study, the wild-type control group exhibited the lowest frequency of seizure attacks, while the model control group showed the highest frequency. The seizure frequency in the model control group was significantly higher than in the wild-type group at both P50 (P = 0.0004; Mean Diff = -7.5, CL95% (−12.110 to −2.891)) and P57 (P = 0.02; Mean Diff = -2.67, CL95% (−5.008 to −0.325)). Furthermore, the frequency of abnormal discharge on EEG and seizure attacks at P50 was significantly decreased in the HMGB1- and IL-1β-prevention and -intervention group compared to the model control group (Figure 4). These findings suggest that targeting HMGB1 and IL-1β can mitigate seizures and abnormal EEG discharges by alleviating the inflammatory response. At P57, the model control mice continued to display a significantly higher frequency of abnormal EEG discharges than the wild-type controls (P = 0.02; Mean Diff = -2.667, CL95% (−5.502 to −0.281)). Overall, the results indicate that HMGB1 and IL-1β have a detrimental effect on epilepsy progression. Interfering with these inflammatory mediators led to a reduction in seizure frequency, highlighting their potential as therapeutic targets for epilepsy treatment.
Discussion
TSC is an autosomal dominant, multisystem neurocutaneous disorder, with all brain cells exhibiting the same underlying genetic mutation. 32 However, findings from preoperative EEG, MRI, and postoperative seizure control confirm that not all brain regions contain cortical tubers, and not all tubers function as epileptogenic tubers (ETs).2,33 This suggests that the two-hit mechanism driving the hyper-epileptogenicity of ETs is likely critical for developing effective strategies to prevent and treat TSC-related seizures. 34 Epigenetic mechanisms, such as DNA methylation and microRNAs, have been implicated in cortical dysplasia. 35 Additionally, IL-1β has been shown to be overexpressed in both patients and animal models with TSC-related epilepsy.36,37 Investigating the biological roles of immune and inflammatory processes in epileptogenesis could facilitate the discovery of novel biomarkers for more effective patient screening. It is therefore essential to deepen our understanding of immune- and inflammation-related molecules involved in epilepsy to identify new therapeutic targets. Many studies have found the involvement of HMGB1-mediated inflammatory pathways in neurological disorders, such as in temporal lobe epilepsy and other focal epilepsies, chronic neuroinflammation plays a significant role in the development and progression of seizures. Similar inflammatory pathways, such as those involving HMGB1 and IL-1β, have been implicated in neurodegenerative diseases like Alzheimer's and Parkinson's, where neuroinflammation is a driving factor in disease progression.11,38 These common inflammatory mechanisms may also influence neuronal hyperexcitability, contributing to seizure generation in epilepsy and exacerbating neurodegenerative processes in other disorders.11,38 Epilepsy remains inadequately controlled with existing therapies, underscoring the need for further research into the mechanisms of HMGB1 in epilepsy.11,12,18
Many researchers have reported a correlation between the inflammatory response and epilepsy.36,37 However, the precise roles of inflammation and HMGB1 require further investigation through in vivo and in vitro experiments to elucidate their complex interactions. Therefore, we performed molecular biological and behavioral verification in animal studies. The over-expression of HMGB1, TLR4, IL-1β, and IL-1R in ETs compared to non-ETs, was proved in wild-type and model control group in mice. Furthermore, the frequency of seizures was lowest in the wild-type control group and highest in the model control group, supporting the high epileptogenicity in ETs and TSC2-deficient mice. Although the TSC2 mutant mouse model provides important biological insights into TSC, it is unable to exhibit obvious cortical tubers, which limits a comprehensive understanding of the human disease.30,39 There are significant differences between mice and humans in cortical development, structure, and response to environmental stimuli. 39 The complexity of human cortical development and tuber formation mechanisms are not fully replicated in mouse models. Species-specific factors may influence cortical tuber formation in mice, making it difficult to accurately reflect human TSC pathology. 40 Additionally, phenotypic responses to TSC2 mutations may vary due to differences in regulatory mechanisms, such as cortical neuron development, synaptic connectivity, and injury repair. 40 While the mouse model cannot fully replicate human cortical tuber formation, it does show abnormal GFAP-immunoreactive astrocyte proliferation and cell size regulation in the neocortex,30,39 along with observable neurological activity related to epilepsy.
Furthermore, our findings revealed that HMGB1 predominantly localizes to the nucleus of neurons in non-ETs, whereas in ETs, it is mainly expressed in the cytoplasm. This suggests a crucial translocation mechanism of HMGB1 in epilepsy, potentially involved in the early stages of epileptogenesis. We propose that epileptic seizures act as key triggers for the translocation of HMGB1, facilitating its release into the cytoplasm. When brain cell injury or death occurs, such as through TSC or cortical dysplasia, HMGB1 is actively released from the intracellular environment into the extracellular space. 41 Moreover, immunofluorescence analysis demonstrated a consistent expression pattern for HMGB1 and IL-1β, suggesting that HMGB1 activates IL-1 signaling within neurons, playing a pivotal role in the initiation and recurrence of seizures. 16 The distinct distribution of HMGB1 between ETs and non-ETs may provide insights into the mechanisms of neuronal damage contributing to epilepsy. HMGB1 may amplify IL-1 signaling via TLR4 and RAGE, activating NF-κB and inflammasome pathways, which upregulate IL-1β and other pro-inflammatory cytokines. 42 This enhances neuronal hyperexcitability and seizure recurrence. Additionally, HMGB1 plays a crucial role in neuroinflammation when released into the extracellular space. Its release and translocation can activate various signaling pathways, including the TLR4/NLRP3/Caspase-1 pathway, which is involved in the activation of IL-1β. 43 The activation of IL-1β, in turn, exacerbates neuroinflammation, which is a key contributor to seizure generation and progression in epilepsy. Understanding the precise mechanisms of HMGB1 translocation in neurons could help identify key regulatory points for therapeutic intervention. Moreover, the HMGB1-IL-1β interaction may create a self-sustaining inflammatory loop, where recurrent seizures drive continuous HMGB1 release, fueling chronic neuroinflammation and epileptic progression.
Our findings demonstrated that a reduction in HMGB1 levels led to a corresponding decrease in TLR4 and IL-1β expression. However, when IL-1β was inhibited, HMGB1 levels also showed a decline. Moreover, the frequency of seizure attacks and abnormal discharges on EEG were significantly reduced in both the prevention and intervention groups targeting HMGB1 or IL-1β, compared to the model control group. These results indicate that early prevention and late intervention are both effective in lowering seizure frequency. Electrophysiological analysis further confirmed that HMGB1 and IL-1β influence neuronal excitability. Based on these observations, we propose that HMGB1 and IL-1β may engage in a positive regulatory loop, with HMGB1 modulating the inflammatory response through IL-1β, thereby contributing to the pathogenesis of epilepsy. When further investigating the potential regulatory role of HMGB1 on TLR4 and IL-1β receptors, several factors need to be considered. First, HMGB1, as an important inflammatory mediator, is known to mediate its effects through various receptors, with TLR4 and the IL-1β receptor being among the key ones. 11 HMGB1 may initiate downstream inflammatory responses, such as the activation of the NF-κB signaling pathway, by binding to these receptors, which is crucial for immune and inflammatory processes. 44 However, our experimental results failed to show any impact of changes in HMGB1 and IL-1β expression on TLR4 and IL-1β receptor levels, which may be attributed to several factors. On one hand, the effects of HMGB1 may be mediated through other receptors or regulatory pathways not examined in this study, rather than solely depending on TLR4 and IL-1β receptors. For example, HMGB1 may interact with other receptors, such as the receptor for advanced glycation end products, 42 to activate alternative signaling pathways, thereby indirectly regulating immune responses and inflammatory processes. Additionally, the interaction between HMGB1 and its receptors may not be limited to changes at the gene expression level, but could also involve the regulation of receptor function through post-translational modifications, which requires further detailed analysis and experiments. Treatment with anti-HMGB1 and anti-IL-1β antibodies demonstrated promising therapeutic effects in the epileptic animal model. Notably, HMGB1-driven inflammation through the IL-1β pathway appears to promote seizure activity.
Limitation
This study had limitations. First, the sample size remains limited due to the low prevalence of TSC-related epilepsy and the relatively small number of patients undergoing resective surgery. The limited sample size could affect the generalizability of the findings and the statistical power of the results. Additionally, the small cohort may not fully capture the heterogeneity of TSC-related epilepsy in terms of severity, comorbidities, and response to treatment. To address this limitation, future research could involve multicenter studies or large-scale cohort studies to increase sample size and improve the diversity of participants. Second, in the animal model, we did not fully investigate the intracellular translocation mechanisms of HMGB1 in epilepsy, which will be the focus of future research.
Future directions
Future research should focus on clarifying the role of HMGB1 and IL-1β in chronic neuroinflammation in TSC-related epilepsy. Investigating the dynamics of HMGB1 translocation and release during epileptogenesis could help identify it as an early biomarker for epilepsy. Disrupting the HMGB1-IL-1β inflammatory loop may also present new therapeutic opportunities. Developing inhibitors to block HMGB1 release or its interaction with downstream pathways could break this self-perpetuating cycle and reduce both the frequency and severity of seizures. Furthermore, exploring early intervention and preventive strategies to regulate HMGB1 expression and function could delay epilepsy onset, slow disease progression, and improve neurodevelopment. Investigating the role of other pro-inflammatory cytokines or transcription factors, and their interactions with excitatory neurotransmitter systems, could enhance our understanding of epilepsy's pathophysiology. These studies may provide new preventive and therapeutic strategies for epilepsy and other neurological disorders with significant clinical potential.
Conclusion
The interference of HMGB1 and IL-1β could significantly reduce the frequency of seizure attacks. Additionally, epileptic seizures serve as key triggers for HMGB1 translocation and its subsequent release into the cytoplasm. It is likely that HMGB1 regulates IL-1β through a specific signaling pathway. Based on the investigation of its anti-inflammatory mechanisms, HMGB1 emerges as a potential biomarker for drug-resistant epilepsy and a promising target for antiepileptic therapies.
Supplemental Material
sj-docx-1-sci-10.1177_00368504251338653 - Supplemental material for HMGB1 mediates inflammation response of epileptogenicity in tuberous sclerosis complex-related epilepsy
Supplemental material, sj-docx-1-sci-10.1177_00368504251338653 for HMGB1 mediates inflammation response of epileptogenicity in tuberous sclerosis complex-related epilepsy by Suhui Kuang, Tinghong Liu, Liu Yuan, Jinshan Xu, Yangshuo Wang, Feng Chen and Shuli Liang in Science Progress
Footnotes
Acknowledgment
The authors would like to show gratitude to the patients and their families for their long-term cooperation. We also appreciate the contribution provided by the following people: Wanshan Xu and Wenyan Zhang in the Beijing Institute of Pediatric.
Ethical considerations
This study was approved by the Ethics Committee of Beijing Children's Hospital, Capital Medical University ([2022]-E-022-Y; 2022–01–25), and written informed consent was obtained from each patient. Care and use of mice were conducted according to an animal protocol approved by SPF (Beijing) Biotechnology Co., Ltd (AWE 2023011101; 2023-01-11).
Author contributions
K.S., L.S. and L.T. writing-original draft, data curation and methodology. L.S. and K.S. conceptualization. Y.L. and X.J. writing- review& editing and data curation. W.Y. and C.F. writing- review& editing and resources. All authors have read and agreed to the version of the manuscript to be submitted.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by Beijing Nature & Science Foundation of China (7202045, SL Liang) and National Nature & Science Foundation of China (82071448, SL Liang). Those funds did not involve the study design, data collection and analysis, interpretation of data and the writing of the report.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.