
Abstract
Background:
Most patients with developmental and epileptic encephalopathy (DEE) have genetic etiology, which has been uncovered with different methods. Although chromosomal microarray analysis (CMA) has been broadly used in patients with DEE, data is still limited.
Methods:
Among 560 children (<18 years) who underwent CMA in our hospital between January 2013 and June 2021, 146 patients with developmental delay and recurrent seizures were screened. Patients with major brain abnormalities, metabolic abnormalities, and specific syndromes were excluded. The rate of rare copy number variants (CNVs) was estimated in total and according to seizure-onset age, relation to first seizure with the diagnosis of developmental delay, epilepsy syndromes, and organ anomalies.
Results:
Among the 110 patients enrolled, the rate of rare CNVs was 16.4%, varying by seizure-onset age: 33.3% in three neonates, 21.2% in 33 infants, 13.3% in 45 early childhood patients, 5.3% in 19 late childhood patients, and 30.0% in 10 adolescents. In relation to the first seizure with the diagnosis of developmental delay, the rates were 3.7%, 22.2%, and 12.5% in “before”, “after”, and “concurrent” subclasses, respectively. The rates of rare CNVs were 16.7% in “other predominantly focal or multifocal epilepsy”, 28.6% in “other predominantly generalized epilepsy (PGE)”, and 15.4% in West syndrome. The rates were 27.8% in minor brain anomalies, 37.5% in facial dysmorphism, and 22.2%, 20.0%, and 57.1% in endocrine, genitourinary and cardiovascular anomalies, respectively.
Conclusion:
The rate of rare CNVs in patients with genetic DEE was 16.4% in total, which was higher in seizures occurring below the infantile period or after the diagnosis of developmental delay, in PGE, and in the presence of facial dysmorphism or cardiovascular anomalies.
Introduction
Developmental and epileptic encephalopathy (DEE) is the new terminology, first introduced in 2017 by the International League Against Epilepsy (ILAE) on the position paper of ILAE for the classification of epilepsies. 1 The suggestion of DEE was initiated based on the existing epilepsy syndromes with genetic etiologies (e.g. Dravet syndrome) that could not be classified into “epileptic encephalopathy” nor “developmental encephalopathy”. 1 Epileptic encephalopathy has severe epileptic activities resulting in developmental and behavioral deterioration, however, with no preexisting developmental disorders, whereas “developmental encephalopathy” has developmental disorders only but with no frequent epileptic activities on an electroencephalogram.1,2 The concept of DEE developed, as genetic insights for epilepsies have markedly improved with the rapid development of accurate and several genetic diagnostic methods over a decade.2–12
Chromosomal microarray analysis (CMA) is one of the genetic diagnostic methods for epilepsy, which has already been tested for patients with unexplained developmental disorders, autism spectrum disorders, and congenital anomalies as a first-tier diagnostic test in clinics.13,14 CMA has been frequently performed in patients with childhood-onset drug-resistant epilepsy, genetic epilepsy with intellectual disability, or genetic DEE.9,10,13,15–21 However, CMA data in different clinical settings are still limited.5–12
The diagnostic yield and cost-effectiveness of CMA are relatively low in epilepsy compared with next-generation sequencing (NGS) or whole exome sequencing (WES).3–6,17,22 Epileptic encephalopathy and pathogenic copy number variants (CNVs) were found in 3%–5% of some studies.20,23 Therefore, more data regarding clinical characteristics of cases with a high positive rate of pathogenic CNVs in CMA need to be investigated to improve the diagnostic yield of CMA. In this study, we investigated the rate of rare CNVs based on seizure-onset age, relation to first seizure with the diagnosis of developmental delay, specific epilepsy syndromes of DEE, and associated anomalies in multiple organs, in patients with genetic DEE but without the known structural, metabolic, or classical syndromic diagnoses.
Methods
The medical records of 560 children (<18 years), who underwent CMA in Chonnam National University Hospital (CNUH) between January 2013 and June 2021, were retrospectively screened. Among them, the 146 patients with developmental delay and recurrent seizures were screened further. There were no self-limited and pharmaco-responsive epilepsy syndromes included. Patients with major brain anomalies (N, 9), hypoxic or traumatic cerebral insults before seizure-onset (N, 7), metabolic abnormalities proven later (N, 4), and specific syndromes diagnosed later by other classical genetic diagnostic methods (N, 6) were excluded. The specific syndromes excluded were fragile X-syndrome (N, 2), Rett syndrome (N, 1), Kabuki syndrome (N, 1), Lowe syndrome (N, 1) and XYY syndrome (N, 1). Cases with insufficient data (N, 10) were also excluded: cases requiring specific methylation tests for genomic imprinting, which was however not performed, were also excluded. On the other hand, patients with marker chromosomes and interstitial deletion or duplication that have to be further evaluated with CMA were included. Finally, the remaining 110 patients who exhibited genetic DEE without structural or metabolic etiology were enrolled in our study (Fig. 1).

Patients (<18 years of age) with developmental and epileptic encephalopathy who underwent chromosomal microarray analysis between January 2013 and June 2021 at Chonnam National University Hospital. *The specific syndromes diagnosed later by other classical genetic diagnostic methods included fragile X-syndrome (N, 2), Rett syndrome (N, 1), Kabuki syndrome (N, 1), Lowe syndrome (N, 1), and XYY syndrome (N, 1).
Study design
The clinical data of patients were thoroughly reviewed, including age, sex, birth history, family history, seizure-onset age, developmental delay onset age, history of seizures and development, and associated anomalies. The results of CMA and other evaluation methods (e.g. brain image studies, electroencephalography, metabolic screening, and genetic tests) were also reviewed. The pathogenicity of variants in CMA was interpreted utilizing the 2011 American College of Medical Genetics and Genomics guidelines. 24 Among the 110 patients enrolled, 96 patients were tested using a single nucleotide polymorphism array with either of CytoScan 750 K array (Affymetrix, Inc., Santa Clara, CA, USA) or CytoScan Dx Assays (Thermo Fisher Scientific, Inc., Waltham, MA, USA) in reference to the human genome reference build 19 (Hg 19), and 14 patients were tested with array comparative genomic hybridization (array CGH, NimbleGen CGX-3 135 K whole-genome array; Roche NimbleGen, Inc., Madison, WI, USA) referring to Hg 18.
The rate of pathogenic and possibly pathogenic variants in CMA related to the developmental disorder or epilepsy was estimated in the 110 patients enrolled.17,24 The rates of rare CNVs were further analyzed according to the different circumstances: 1) the subgroups of seizure-onset age; 2) relation to first seizure with the diagnosis of developmental delay; 3) specific epilepsy syndromes; and 4) organ systems with anomalies.
The subgroups of age were divided into five: neonatal period (<1 month of age), infantile period (1–11 months of age), early childhood (1–5 years), late childhood (6–10 years), and adolescent period (11–17 years). The subclassification of relation to the first seizure with the diagnosis of developmental delay was 1) before, 2) after, and 3) concurrent. Specific epilepsy syndromes were classified as follows: 1) early infantile epileptic encephalopathy (EIEE, Ohtahara syndrome); 2) early myoclonic encephalopathy (EME); 3) epilepsy of infancy with migrating focal seizures (EIMFS); 4) early onset epileptic encephalopathy (EOEE, before 3 months of age); 5) Dravet syndrome (DS); 6) West syndrome (WS); 7) epilepsy with myoclonic-atonic seizures (EMA); 8) Lennox–Gastaut syndrome (LGS); 9) epilepsy-aphasia spectrum (EAS); 10) other predominantly myoclonic epilepsy (PME); 11) other predominantly focal or multifocal epilepsy (PFE); and 12) other predominantly generalized epilepsy (PGE).2,7 Anomalies were categorized according to organ systems: cardiovascular system; pulmonary system; genitourinary system; gastrointestinal system; osteoskeletal system; endocrine system; ophthalmologic system; ears, nose, and throat; and facial dysmorphism.
The rare CNVs were further summarized in each patient. For the variants subjected to genomic imprinting, the results of the methylation test of specific genes within the variant sites were further reviewed. Our study was approved by the ethics committee of CNUH (IRB approval number: CNUH-2021-351). The requirement for informed consent was waived in this retrospective study.
Results
Rates of rare CNVs according to seizure-onset age and relation to first seizure with the diagnosis of developmental delay
In a total of 110 patients with genetic DEE (male N, 73), the seizure-onset age was 48.4 ± 52.6 months (mean ± standard deviation). The majority of the patients (70.9%; N, 78) experienced their first seizures in infancy and early childhood (1 month–5 years). In 65.5% of patients (N, 72), they presented their first seizures after developmental delay onset.
The rate of rare CNVs was 16.4% (N, 18) in total, which varied according to seizure-onset age groups: 33.3% (1/3) in neonates, 21.2% (7/33) in infants, 13.3% (6/45) in early childhood patients, 5.3% (1/19) in late childhood patients, and 30.0% (3/10) in adolescents. The rates of rare CNVs according to relation to the first seizure with the diagnosis of developmental delay were 3.7% (1/27) in “before,” 22.2% (16/72) in “after,” and 12.5% (1/8) in the “concurrent” group (Table 1).
Clinical overview and rates of rare CNVs in patients with genetic DEE (total N, 110).
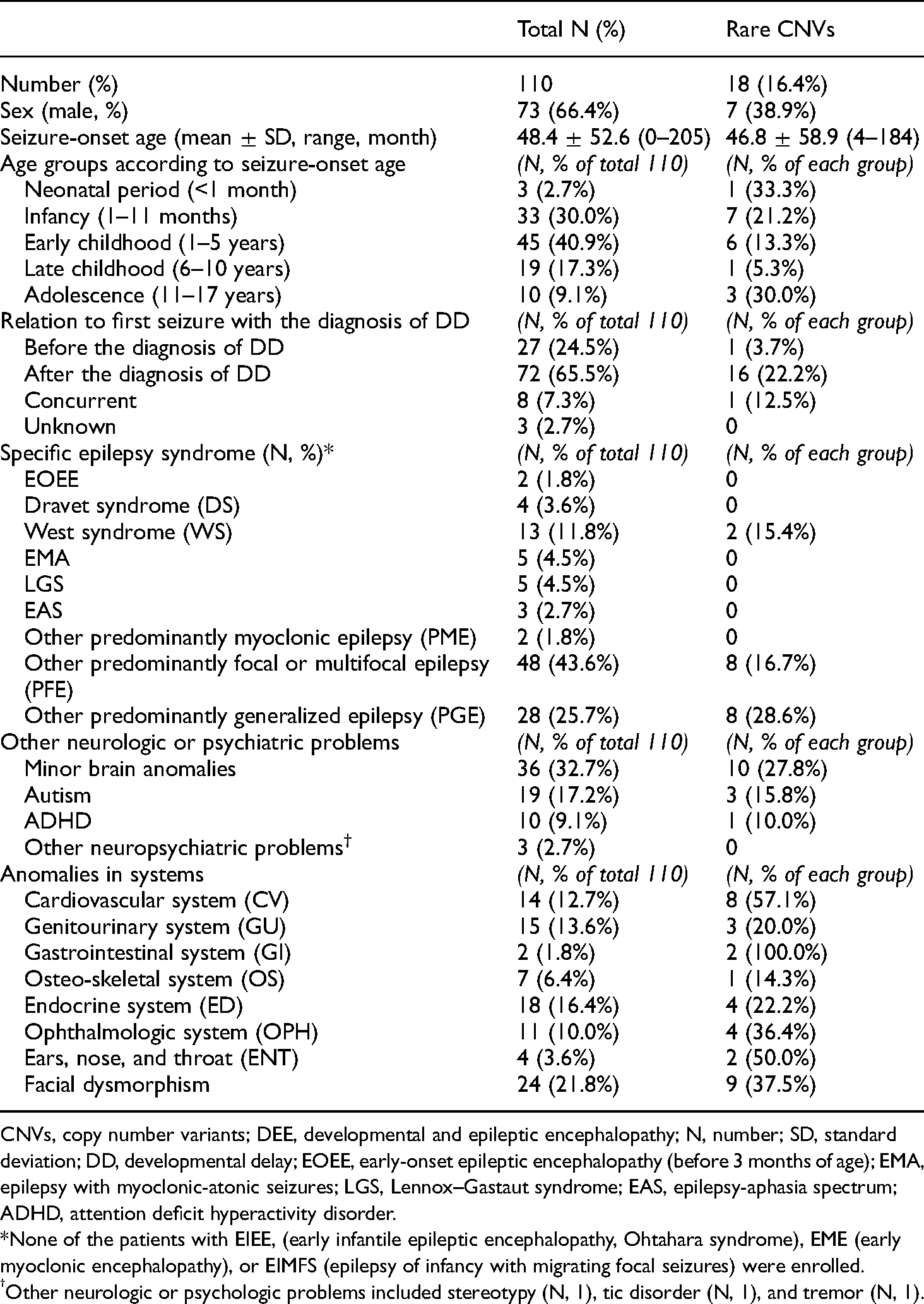
CNVs, copy number variants; DEE, developmental and epileptic encephalopathy; N, number; SD, standard deviation; DD, developmental delay; EOEE, early-onset epileptic encephalopathy (before 3 months of age); EMA, epilepsy with myoclonic-atonic seizures; LGS, Lennox–Gastaut syndrome; EAS, epilepsy-aphasia spectrum; ADHD, attention deficit hyperactivity disorder.
*None of the patients with EIEE, (early infantile epileptic encephalopathy, Ohtahara syndrome), EME (early myoclonic encephalopathy), or EIMFS (epilepsy of infancy with migrating focal seizures) were enrolled.
Other neurologic or psychologic problems included stereotypy (N, 1), tic disorder (N, 1), and tremor (N, 1).
Rates of rare CNVs in each epilepsy syndrome
Among epilepsy or epilepsy syndromes found in our enrolled patients, other PFE (43.6%; N, 48), other PGE (25.7%; N, 28), and WS (11.8%; N, 13) were common. For severe epilepsy syndromes of EIEE, EME, or EIMFS that occur in the neonatal period or in early infancy, no patients were enrolled in the current study.
The rates of rare CNVs according to specific epilepsy syndrome were 16.7% (8/48) in PFE, 28.6% (8/28) in PGE, and 15.4% (2/13) in WS. No abnormal variants were found in other epilepsy syndromes (Table 1).
Rates of rare CMA in other neurologic or psychiatric problems
In all patients with genetic DEE, minor brain anomalies were detected in 36 patients (32.7%), which included ventricular anomaly (N, 13), corpus callosal dysgenesis (N, 12), mild brain atrophy (N, 7), arachnoid cyst (N, 6), vascular malformation (N, 4), dysmorphic basal ganglia or thalami (N, 2), subtle gyrus anomaly (N, 2), hippocampal sclerosis (N, 2), and others. Autism (17.2%; N, 19), attention deficit hyperactivity disorder (ADHD) (9.1%; N, 10), stereotypy (N, 1), tic disorder (N, 1), and tremor (N, 1) were also noted.
The rates of rare CNVs were 27.8% (10/36) in patients with minor brain anomalies, 15.8% (3/19) in autism, and 10.1% (1/10) in ADHD (Table 1).
Rates of rare CNVs in each systematic anomaly
In patients with genetic DEE, facial dysmorphism was the most frequently observed (21.8%; N, 24), followed by endocrinologic anomalies (16.4%; N, 18), including short stature (N, 12), hypothyroidism (N, 3), and others. Genitourinary anomalies were noted in 15 patients (13.6%), consisting of cryptorchidism (N, 7), inguinal hernia (N, 2), vesicoureteral reflux (N, 2), and others. Cardiovascular anomalies were detected in 14 patients (12.7%): atrial septal defect (N, 6), ventricular septal defect (N, 4), patent ductus arteriosus (N, 4), bicuspid aortic valve (N, 2), and others. Ophthalmologic anomalies were observed in 11 patients (10.0%), whereas osteoskeletal, otorhinolaryngologic, or gastrointestinal anomalies were noted below 10% each. No pulmonary anomalies were noted.
The rates of rare CNVs according to associated anomalies found in >10% of patients were 37.5% (9/24) in “facial dysmorphism,” 22.2% (4/18) in the “endocrinologic anomalies”, 20.0% (3/15) in the “genitourinary anomalies,” and 57.1% (8/14) in the “cardiovascular anomalies”, respectively (Table 1).
Pathogenic or possibly pathogenic variants of CMA in each patient with genetic DEE (n, 17)
In 18 patients, 21 abnormal findings in CMA were detected, as three patients had two different abnormalities. There were no data on family segregation in the clinical chart of any patient. Among the 21 abnormalities, most were deletions (66.7%; N, 14). Others were duplication (N, 3), triplication (N, 3), and loss of heterozygosity (4.8%, N, 1). Among the three patients with triplication, two had a marker chromosome (patients 12 and 17). The arms of chromosomes with abnormalities were 1p (N, 2), 1q (N, 1), 3p (N, 1), 3q (N, 2), 4p and q (N, 1), 9q (N, 1),10q (N, 3), 14q (N, 2), 15q (N, 3), 16p (N, 2), 18q (N, 2), and Xq (N, 1). Therefore, 10q (two with 10q 26.3 deletion and one with 10q23.1-q23.2 deletion) and 15q (two with Prader–Willi syndrome and one with Angelman syndrome) were the most commonly reported sites (14.3% for each). There were two FOXG1-related disorders: one female with 14q12 deletion (atypical Rett syndrome) and one female with 14q11.2q13.1 triplication (FOXG1 duplication syndrome). There was one male with Xq28 duplication (MECP2 duplication syndrome). “Patient 4” had 18q21.2 microdeletion, in which the chromosomal site included TCF4 of the gene related to Pitt–Hopkins syndrome (Tables 2 and 3).
Findings of rare CNVs in genetic DEE patients without structural or metabolic etiology.

CNVs, copy number variants; DEE, developmental and epileptic encephalopathy; DD, developmental delay; CMA, chromosomal microarray analysis; F, female; M, male; Other PGE, other predominantly generalized epilepsy; Other PFE, other predominantly focal or multifocal epilepsy; WS, West syndrome; dup, duplication; del, deletion; trp, triplication; LOH, loss of heterozygosity.
*Methylation test of SNRPN gene showed maternal homologue SNRPN methylation pattern, which is consistent with Prader–Willi syndrome.
Methylation test of SNRPN gene showed paternal homologue SNRPN methylation patterns, which is consistent with Angelman syndrome.
Methylation specific-multiplex ligation-dependent probe amplification (MLPA) showed an increase of the dosage quotient median (0.765 for SNRPN-CpG island; 0.74 for MAGEL2-CpG island), which are suggestive of three maternal imprinted alleles with one paternal imprinted allele as the dosage quotients are “1.0” for Prader–Willi syndrome, “0.5” for normal and “0” for Angelman syndrome.
Associated anomalies and other neuropsychiatric problems in genetic DEE patients with rare CNVs.

DEE, developmental and epileptic encephalopathy; CNVs, copy number variants; del, deletion; dup, duplication; trp, triplication; LOH, loss of heterozygosity; PDA, patent ductus arteriosus; ASD, atrial septal defect; VSD, ventricular septal defect; DM, diabetes mellitus; PFO, patent foramen ovale; ADHD, attention deficit hyperactivity disorder.
*Methylation test of SNRPN gene showed maternal homologue SNRPN methylation pattern, which is consistent with Prader–Willi syndrome.
Methylation test of SNRPN gene showed paternal homologue SNRPN methylation patterns, which is consistent with Angelman syndrome.
Methylation specific-multiplex ligation-dependent probe amplification (MLPA) showed an increase in the dosage quotient median (0.765 for SNRPN-CpG island; 0.74 for MAGEL2-CpG island), which are suggestive of three maternal imprinted alleles with one paternal imprinted allele as the dosage quotients are “1.0” for Prader–Willi syndrome, “0.5” for normal and “0” for Angelman syndrome.
Discussion
There are specific epilepsies or epilepsy syndromes included in the DEE, in which seizures started at different ages.2,7 EIEE, EME, EIMFS, and EOEE are usually observed before 3 months of age. WS and DS are reported before 1 year of age, while EMA, LGS, and EAS are recognized beyond infancy. “Other PME” and “other PFE” are seen at any age.2,7 For each specific epilepsy syndrome, there are some reports for the frequencies of rare CNVs. 23 For infantile spasm, Michaud found that 6.8% of 44 patients had rare CNVs in CMA: a 2q21.3-q22.2 deletion, a 16p11.2 duplication, and a 15q11.1q13.1 tetrasomy. 25 Mefford et al. also reported that rare CNVs were found in 6.8% of 44 patients with infantile spasm. 23 In our report including 13 patients with WS, two patients (15.4%) had rare CNVs: a 1p36.33-p36.32 deletion and a 14q11.2q13.1 triplication. For other epilepsy syndromes, EIEE, EME, or EIMFS was not enrolled in our study, and no CNV was found in small numbers of patients with EOEE, DS, EMA, LGS, or EAS. In another study, rare CNVs were found to be 9.1% in EIMFS (N, 11), 5.3% in DS (N, 19), 7.8% in EMA (N, 77), and 10.3% in EAS (N, 29), whereas rare CNVs were not detected in 20 patients with LGS. 23
In our DEE data, there were patients with more bilateral tonic-clonic seizures than focal seizures not classified into the specific epilepsy syndromes; these patients were classified under the “other PGE group.” Among our 28 patients with other PGE of different ages, eight patients (28.6%) had rare CNVs. Mefford et al. also mentioned “severe idiopathic generalized epilepsy of infancy” (N, 15) in their CNV study, and reported two patients (13.3%) with rare CNVs: a 16p11.2 microdeletion and a 2q35 microduplication. 26 Notably in our study, 16p11.2 microdeletion was found in two patients with other PFE during infancy, not with other PGE. In our study including 48 patients with PFE, rare CNVs were found in 16.7% (N, 8) of patients with PFE. In another study by Mefford et al., rare CNVs were found in 23.5% of 17 patients with focal epilepsy and regression. 23
The microdeletion of 15q13.3 encompassing the CHRNA7 has been reported to contribute to the phenotype of genetic generalized epilepsy.23,27 Microdeletion restricted to 15q11.2, 15q13.3, or 16p13.11 have been reported usually in genetic generalized epilepsy, but rarely in epileptic encephalopathy or DEE.10,16,23,26–28 In our report, one patient with 15q11.2-q13.3 triplication (three maternal imprinted alleles vs. one paternal allele) had PGE. Considering this case, the role of 15q13.3 or 15q11.2 microduplication on generalized seizures also needs to be further evaluated.
The rate of rare CNVs in genetic DEE without major brain anomalies was 16.4% of 110 patients in our study; however, data to date is still limited, as the definition of DEE has been suggested recently and the major cortical malformations were included in most CNV studies for epileptic encephalopathy. Furthermore, the rare CNV rates by seizure-onset age, by relation to the first seizure with the diagnosis of developmental delay, and by associated congenital abnormalities have been rarely reported. In our report, the rare CNVs were found as follows: 21.2% in infants (N, 33), 13.3% in early childhood patients (N, 45), and 5.3% in late childhood patients (N, 19). Although the rate of rare CNVs was about 33% in three neonates and 30% in 10 adolescents, they need to be reproduced in sufficient numbers of patients. The rates of rare CNVs according to relation to first seizure with the diagnosis of developmental delays were 22.2% in “first seizure after the diagnosis of developmental delay” (N, 72), 12.5% in “concurrent” (N, 8), and 3.7% in “before” (N, 27). A CMA study performed by Hrabik et al. on 147 children with epilepsy showed that rare CNVs were significantly higher in patients with musculoskeletal or cardiovascular malformation. 21 In our study, the rare CNVs were also frequently detected in patients with cardiovascular anomalies (57.1% of total 14 patients) and facial dysmorphism (37.5% of total 24 patients). However, in seven patients with osteoskeletal anomalies, only one had rare CNV (14.3%). In our study, patients with minor brain anomalies (N, 36) were included, among whom, the rate of rare CNVs was 27.8% (N, 10).
This study has some limitations. First, the available data are inadequate to conduct a familial segregation study. Therefore, all rare CNVs we reported were sorted as “possibly pathogenic variants.” This limitation is supposed because this is a retrospective study. Second, extensive statistical analysis with statistical significance evaluations was not possible because the subgroups were too small to draw a comparison. In future studies, anterograde trio studies involving a larger number of patients are warranted.
Conclusion
To improve the diagnostic yield of CMA in patients with genetic DEE but without the known structural, metabolic, or classical syndromic diagnoses, we suggest that CMA should be recommended more in cases as mentioned below: 1) with the first seizure occurring <1 year of age, 2) with the first seizure being presented after the diagnosis of developmental delay, 3) with PGE, and 4) with cardiovascular anomalies or facial dysmorphism.
Footnotes
Authors’ contributions
Study concept and design: Kim YO. Acquisition of data: Kim YO, Lee SH, and Kim BR. Analysis and interpretation of data: Kim YO, Lee SH, and Kim BR. Drafting of the manuscript: Kim YO and Lee SH. Critical revision of the manuscript: Kim YO.
Ethical approval
This study was approved by the Human Research Ethics Committee of Chonnam National University Hospital (Gwangju, South Korea; IRB approval number: CNUH-2021-351). The requirement for informed consent was waived in this retrospective study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.