
Abstract
Objective
Deep vein thrombosis (DVT) is a global health issue caused by abnormal clotting in deep veins, which can lead to serious complications such as pulmonary embolism. This study is the first to validate the regulatory effect of miR-223-3p on the NLRP3 inflammasome in a mouse model of DVT, expanding its potential therapeutic value in venous thrombosis-associated inflammation.
Methods
MicroRNA sequencing and quantitative real-time polymerase chain reaction (qRT–PCR) were conducted to assess miRNA expression in a DVT mouse model. The downstream target of miR-223-3p, NLRP3, was identified using miRNA target prediction databases and validated by qRT–PCR. Human umbilical vein endothelial cells (HUVECs) and a DVT mouse model were used to explore the functional relationship between miR-223-3p and Nlrp3.
Results
The expression of miR-223-3p and Nlrp3 was significantly increased in the vein walls of mice with DVT. The tail vein injection of agomiR-223-3p reduced thrombus formation and downregulated the expression of Nlrp3, interleukin 6 (Il-6), interleukin 1 beta (IL-1beta) and Icam-1. In vitro, miR-223-3p overexpression reduced the expression of NLRP3, Il-6, IL-1beta and ICAM-1, whereas NLRP3 overexpression antagonized these effects. Additionally, miR-223-3p enhanced the viability and migration of LPS-stimulated HUVECs by reducing NLRP3 expression.
Conclusions
Our findings suggest that miR-223-3p may play a role in alleviating inflammation and reducing the thrombus burden in mice with DVT by downregulating Nlrp3 expression, supporting its potential as a therapeutic target for DVT.
Keywords
Introduction
Deep vein thrombosis (DVT), which refers to blood clots that form within deep veins, can be asymptomatic or accompanied by symptoms such as pain and swelling in the limbs. However, when clots detach and deposit in pulmonary vessels, pulmonary embolism can occur. 1 DVT and its primary complication, pulmonary embolism, are important global health problems. While therapies such as heparin and direct oral anticoagulants (DOACs) are commonly used to manage DVT, the associated risk of bleeding cannot be completely mitigated. 2 The traditional Virchow's triad, i.e. altered blood flow, endothelial injury, and blood hypercoagulability, are insufficient to fully explain the complex mechanisms of DVT formation, 3 emphasizing the need for more comprehensive studies into the molecular underpinnings of DVT to uncover new therapeutic targets.
MicroRNAs (miRNAs) are short noncoding RNAs approximately 19–25 nucleotides in length that regulate gene expression by binding to the 3′ untranslated region (3′UTR) of target genes. 4 Recent studies have implicated miR-223-3p in cardiovascular diseases, where it is thought to be involved in endothelial dysfunction and inflammation. 5 For example, miR-223-3p levels are elevated in atherosclerotic plaques and the serum of patients with atherosclerosis, serving as a potential marker of plaque instability. 6 Elevated miR-223-3p levels have also been observed in the plasma of DVT patients, making it a promising biomarker for DVT diagnosis when combined with D-dimer and miR-125a-5p. 7 Additionally, septic platelets release platelet-derived microparticles (PMPs) carrying miR-223, which can downregulate ICAM-1 expression in endothelial cells, potentially offering protection against excessive sepsis-induced vascular inflammation. 8
Sterile inflammation is central to DVT pathophysiology, contributing to venous endothelial disruption and promoting thrombosis formation. 9 The NLRP3 inflammasome is a key inflammatory regulator involved in various vascular-related diseases, including DVT. 10 NLRP3 inflammasome activation triggers the release of proinflammatory cytokines such as IL-1beta and IL-18, which drive endothelial dysfunction and thrombosis formation. 11 NLRP3 also plays a role in inducing IL-6, which exacerbates inflammatory damage to cells. 12 Given its central role in inflammatory pathways, NLRP3 has been identified as a promising therapeutic target for mitigating thrombotic processes.
miR-223-3p is known to target NLRP3, inhibiting its activation and reducing pyroptosis in human umbilical vein endothelial cells (HUVECs). 13 Studies on thromboangiitis obliterans (TAO) and atherosclerosis have demonstrated the ability of miR-223 to regulate NLRP3, reduce local inflammation, and mitigate thrombosis,14,15 but its specific role in venous inflammation, particularly in DVT, has not been systematically explored.
Although miR-223-3p inhibits NLRP3 under inflammatory conditions, its role under thromboinflammatory stress, such as in DVT, remains unclear. In atherosclerosis, miR-223-3p protects against endothelial dysfunction, but in DVT, 5 persistent inflammation may override its inhibitory effect, leading to sustained NLRP3 activation. A similar paradox is observed in acute myocardial infarction and preeclampsia (PE),16,17 where miR-223-3p is upregulated but is insufficient to suppress NLRP3 via dominant inflammatory pathways. Unlike arterial thrombosis, which is driven by lipid deposition, venous thrombosis is linked primarily to sterile inflammation, endothelial dysfunction, and platelet activation, 9 yet the regulatory role of miR-223-3p in this context remains unexplored.
Using bioinformatics and experimental approaches, including miRNA mimetics, we confirmed the role of miR-223-3p in NLRP3 regulation and established its potential as a therapeutic target for DVT. These findings lay the groundwork for future miRNA-based interventions in thromboinflammatory disorders.
In this study, we aimed to investigate the role of miR-223-3p in DVT, hypothesizing that miR-223-3p regulates NLRP3-mediated inflammation in the vascular endothelium, thereby inhibiting thrombosis development. While previous studies have explored miR-223-3p in other vascular diseases, our research uniquely focuses on DVT, providing the first in vivo evidence of the regulatory effect of miR-223-3p on NLRP3 in this specific context.
Although miR-223-3p has been shown to suppress NLRP3 in atherosclerosis and other inflammatory models, its role in venous thromboinflammation remains unclear. Here, we provide the first in vivo evidence that miR-223-3p directly regulates NLRP3 in a DVT model. Unlike previous studies that have focused on arterial thrombosis or systemic inflammation, we reveal a compensatory upregulation mechanism in which the level of miR-223-3p increases in response to DVT-associated inflammation but remains insufficient to fully suppress NLRP3 through dominant proinflammatory pathways.
Methods
DVT mouse model and treatment
The reporting of this study conforms to the ARRIVE 2.0 guidelines, 18 and the experimental procedures and animal care adhered to the guidelines set forth in the Guide for the Care and Use of Laboratory Animals, 8th Edition. 19 Ten-week-old male C57BL/6J mice (weight, 25 ± 1 g) were purchased from the Experimental Animal Department of Kunming Medical University (Kunming, China). The mice were housed under standard laboratory conditions at a temperature of 22 ± 2°C, a relative humidity of 55 ± 10%, and a 12-h light-dark cycle. The mice were randomly allocated into five groups: Blank group (n = 10), DVT group (n = 17), DVT NC group (n = 10), DVT agomir NC group (n = 10), and DVT agomiR-223-3p group (n = 10). Agomir NC or agomiR-223-3p (RiboBio, Guangzhou, China) was administered via a tail vein injection at a dose of 20 nmol/mouse in 200 μL of saline. This dosage was selected based on commonly used concentrations in miRNA-based in vivo studies, 20 where systemic administration typically ranges from 5–20 nmol per injection. The DVT group underwent inferior vena cava (IVC) stenosis modeling, with 5-0 silk sutures placed below the renal veins using a 30 G insulin needle as a spacer. After 24 h, all mice were sacrificed by cervical dislocation, and thrombus formation was assessed.
Animals were excluded if they exhibited severe postoperative complications, failed tail vein injections, or did not develop thrombi following IVC stenosis. Thrombus formation was used as a quality control criterion to confirm successful modeling and was not treated as an outcome measure.
In the agomiR-223-3p group, animals that failed to form thrombi could have represented either modeling failure or potential miR-223-3p–mediated inhibition of thrombogenesis. However, due to the lack of additional confirmatory evidence (e.g. molecular or histological markers), these animals were conservatively excluded to avoid misinterpretation. The same exclusion criteria were uniformly applied to all experimental groups receiving agomir treatment. Based on these criteria, seven mice in the DVT group were excluded (1 due to postoperative complication, 6 due to failure of thrombus formation); four in the DVT NC group (1 injection failure, 3 failed thrombus formation); five in the DVT agomir NC group (2 injection failures, 3 failed thrombus formation); and four in the DVT agomiR-223-3p group (1 injection failure, 3 failed thrombus formation). The Blank group had no exclusions. The final numbers of animals analyzed in each group were as follows: Blank group (n = 10), DVT group (n = 10), DVT NC group (n = 6), DVT agomir NC group (n = 5), and DVT agomiR-223-3p group (n = 6).
MicroRNA sequencing
The extraction process was performed in strict accordance with the manufacturer's instructions and was performed on ice to ensure the integrity of the RNA. Total RNA was extracted from the thrombi using TRIzol reagent. An RNA library was generated according to Illumina's RNA-seq protocol. The RNA integrity was determined using an Agilent 2100 Bioanalyzer, and RNAs with a value ≥ 7.0 were used for library construction. Total RNA samples were reverse transcribed and amplified by PCR. The samples were sequenced using the Illumina SE 50 platform and normalized to the criteria padj < 0.05 and |log2(fold change) | > 1 to obtain differential miRNA profiles. Clustering was performed with log10 (TPM+1) values using the R package DESeq2 (3.20). Red and blue colors indicate high- and low-expression miRNAs, respectively.
Quantitative real-time polymerase chain reaction (qrt–PCR)
Total RNA was extracted with TRIzol reagent at 4°C. Reverse transcription of miRNA and mRNA from 1 μg of total RNA was performed using a first-strand cDNA synthesis kit and a PrimeScript RT kit (RiboBio C10712-2), respectively, and the resulting products were subsequently used as templates for PCR amplification. The thermocycling conditions were as follows: 95°C for 10 min (1 cycle) for initial denaturation, 95°C for 5 s for subsequent denaturation, and 60°C or 72°C for 60 s (40 cycles) for annealing and extension. The miRNA and gene expression levels were normalized to the levels of U6 and β-actin, respectively, according to the relative quantification method (2-ΔΔCt method). For miRNA analysis, the PCR forward primers used were obtained from RiboBio Biotech (miRACM001-12), and the reverse primers used were obtained from a miDETECT-A-TrackTM-miRNA-qRT–PCR Kit (RiboBio Biotech). The mRNA primers used were designed with Primer Premier 5.0, and the primer sequences are shown in Table 1.
Primer sequence.

Bioinformatics analysis
The bioinformatics databases ‘TargetScan’ (https://www.targetscan.org/), ‘miRDB’ (https://mirdb.org/) and ‘miRWalk’ (http://mirwalk.umm.uni-heidelberg.de/) were used to predict the target genes of miRNAs, and the results were subsequently compared with the mRNA-seq results from our previous studies. 21 A Venn diagram was generated to screen out common differential genes (cogenes) and obtain a pooled spectrum of common differential target genes (https://bioinfogp.cnb.csic.es/tools/venny/index.html). The R package clusterProfiler 4.0 was used to test the statistical enrichment of the mRNA-seq results in the KEGG pathways (http://www.genome.jp/kegg/). The samples were normalized to |log2(fold change)| > 1, false discovery rate < 0.05 and padj < 0.05.
Construction of an overexpression plasmid for the human NLRP3 gene
The complete coding sequence (CDS) of human NLRP3 was obtained from GenBank. The following primer sequences were designed via Primer Premier 5.0: NLRP3 (3105 bp) forward, 5'-GGTGGAATTCGAAGTATACCTCGAGGGCAGCTTCGAAG-3’ and reverse, 5'- CGATCGCAGATCCTTGGATCCGTCATGGTCTTTGTAGTC-3’. The human NLRP3 gene was amplified by polymerase chain reaction (PCR), after which the target fragments were detected by agarose gel electrophoresis and subsequently recovered. The fragments and the plasmid vector PGMLV-CMV-Ubc-3xflag-EmGFP-IRES-Puro (Genechem) were subsequently ligated via seamless cloning, after which the concentrations of the recovered vector and target fragments were determined. Finally, the ligated products were transformed into E. coli, and the results were identified via RNA sequencing.
HUVEC culture
Primary HUVECs were purchased from the BeNa Culture Collection (BNCC342438) and cultured in endothelial cell medium at 37 °C in a humidified atmosphere with 5% CO2. The cells were divided into the following groups: I. the normal cell group, II. the miR-223-3p-mimic-NC group, III. the miR-223-3p-mimic group, IV. the empty vector control group (transfected with the plasmid PGMLV-CMV-MCS-EF1-ZsGreen1-T2A-Puro), V. the NLRP3 overexpression group, VI. the miR-223-3p-mimic and empty vector control groups, VII. the miR-223-3p-mimic-NC and empty vector control groups, VIII. the miR-223-3p-mimic and NLRP3 overexpression groups, IX. the miR-223-3p-inhibitor-NC group, and X. the miR-223-3p-inhibitor group. HUVECs were incubated with lipopolysaccharide (LPS) (100 ng/ml) for 24 h, after which chemosynthetic miR-223-3p mimics, inhibitors or the corresponding negative controls (RiboBio Biotech) and the NLRP3 overexpression plasmid were transfected into the cells at a final oligonucleotide concentration of 100 nmol/L with Lipofectamine 3000 (Invitrogen). HUVECs and supernatants from each group were incubated at 37 °C in a 5% CO2 atmosphere for 48 h after transfection.
Western blot analysis
Total protein was extracted from the vein walls and cells with radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitors on ice for 30 min, after which the supernatant was obtained by centrifugation at 4 °C and 12,000 r/min for 15 min. Equal amounts of protein were loaded onto SDS–PAGE gels and transferred to a PVDF membrane. The membrane was blocked with 5% skim milk at room temperature for 1 h and incubated with primary antibodies at 4 °C overnight. The primary antibodies used were as follows: NLRP3 (Bioss, bs-10021R, 1:2000), IL-6 (Bioss, bs-4587R, 1:1000), IL-1beta (Bioss, bs-0812R, 1:1000), and ICAM-1 (Bioss, bs-0608R, 1:2000). β-Actin (Zsbio, TA-09, 1:2000) and GAPDH (Bioss, bs-10900R, 1:4000) served as internal controls. After primary antibody incubation, the membranes were incubated with secondary antibodies (1:4000). The membranes were then incubated with HRP-conjugated goat anti-mouse (Abmart, M21001L) and goat anti-rabbit (Abmart, M21002L) secondary antibodies. Protein band signals were visualized using an enhanced chemiluminescence (ECL) detection kit (NCM Biotech, P10100) and analyzed with ImageJ software.
Enzyme-Linked immunosorbent assay (ELISA)
HUVECs were seeded into a 96-well plate at a density of 1 × 104 cells/mL. The following day, the cells were transfected to produce the following six groups: I. the normal cell group, II. the miR-223-3p-mimic-NC group, III. the miR-223-3p-mimic group, IV. the empty vector control group (transfected with the plasmid PGMLV-CMV-MCS-EF1-ZsGreen1-T2A-Puro), V. the NLRP3 overexpression group; and VIII. the miR-223-3p mimic + NLRP3 overexpression group. After transfection, the HUVECs were incubated with lipopolysaccharide (LPS) (100 ng/ml) for 24 h. The culture supernatant was then collected from the HUVECs, and the protein expression levels of IL-6, IL-1beta, and TNF-α were detected by ELISA using human IL-6, IL-1beta, and TNF-α ELISA kits (Elabscience E-EL-H6156, E-EL-H0149, and E-EL-H0109) according to the manufacturer's instructions. The cells were seeded into a 12-well plate, and the supernatant was collected after 48 h of culture, after which the levels of IL-6, IL-1beta, and TNF-α in the cells were determined. OD values were measured at an excitation wavelength of 450 nm, and the concentrations of IL-6, IL-1beta, and TNF-α in the cell supernatant were calculated using standard curves.
CCK-8 assay
The treated cells were suspended at a concentration of 1 × 104 cells/mL, seeded into a 96-well plate and allowed to adhere overnight. The following day, the same groups of cells were transfected as described for the ELISA experiments. After 48 h, cell viability was measured via a CCK-8 kit (DOJINDO, CK04) by adding 10 μL of CCK-8 solution to each well. The OD value at 450 nm was measured using a microplate reader, and a growth curve was plotted according to the OD values.
Migration assays
Cell migration was measured by a Transwell assay. HUVECs were seeded in 12-well plates at a density of 3 × 104 cells/well. After 24 h of incubation, the cells were transfected according to their group, as in the ELISA experiment. Once the cells had formed a confluent monolayer, a scratch wound was created with a 10 μL pipette tip. The suspended cells were removed by washing with PBS, and 100 μL of basal medium was added to each well. After 0 and 24 h of incubation, the migrated cells were observed and counted under an inverted microscope.
Statistical analysis
All the experiments were repeated independently at least three times. GraphPad Prism 9.0 (GraphPad Software, CA, USA) was used for the statistical analyses. Before applying parametric tests, the Shapiro-Wilk test was used to assess the normality of the data distribution, and Levene's test was performed to confirm the homogeneity of variance across groups. Two-tailed Student's t-tests were used to compare the data between two groups When the normality and equal variance assumptions were met. If the variance was unequal, Welch's t-test was applied. Comparisons of multigroup data were performed using one-way analysis of variance (ANOVA) followed by Tukey's post hoc correction, provided that the assumptions for parametric tests were satisfied. The data are presented as the means ± SDs, and P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
MiR-223-3p is upregulated in DVT mice
To identify miRNAs involved in DVT, we compared the global miRNA expression profiles in the DVT group (n = 3) with those in the blank group (n = 3) using microRNA sequencing analysis. In all, 83 dysregulated miRNAs were identified, including 60 upregulated and 23 downregulated miRNAs (P < 0.05) (Fig. 1A). Among these, 10 candidate miRNAs were selected according to more stringent criteria (P < 0.01 and read count ≥ 100). Real-time quantitative PCR (qPCR) revealed that miR-128-3p, miR-328-3p, and miR-223-3p were significantly upregulated in the DVT group compared with the blank group, whereas miR-455-5p was downregulated (P < 0.05). The expression levels of miR-381-3p, miR-145a-5p, miR-143-5p, miR-881-3p, miR-133a-3p, and miR-222-3p were not significantly different between the groups (P > 0.05) (Fig. 1B). Given the relatively low expression levels of miR-328-3p and miR-455-5p, and despite the high expression of both miR-128-3p and miR-223-3p in the DVT group, we focused on miR-223-3p because of its well-documented role in cardiovascular and inflammatory diseases, which indicates its significant role in regulating inflammation and vascular function. 5 Thus, the high expression of miR-223-3p in DVT and its important role in related diseases prompted us to further investigate its specific mechanism in DVT.
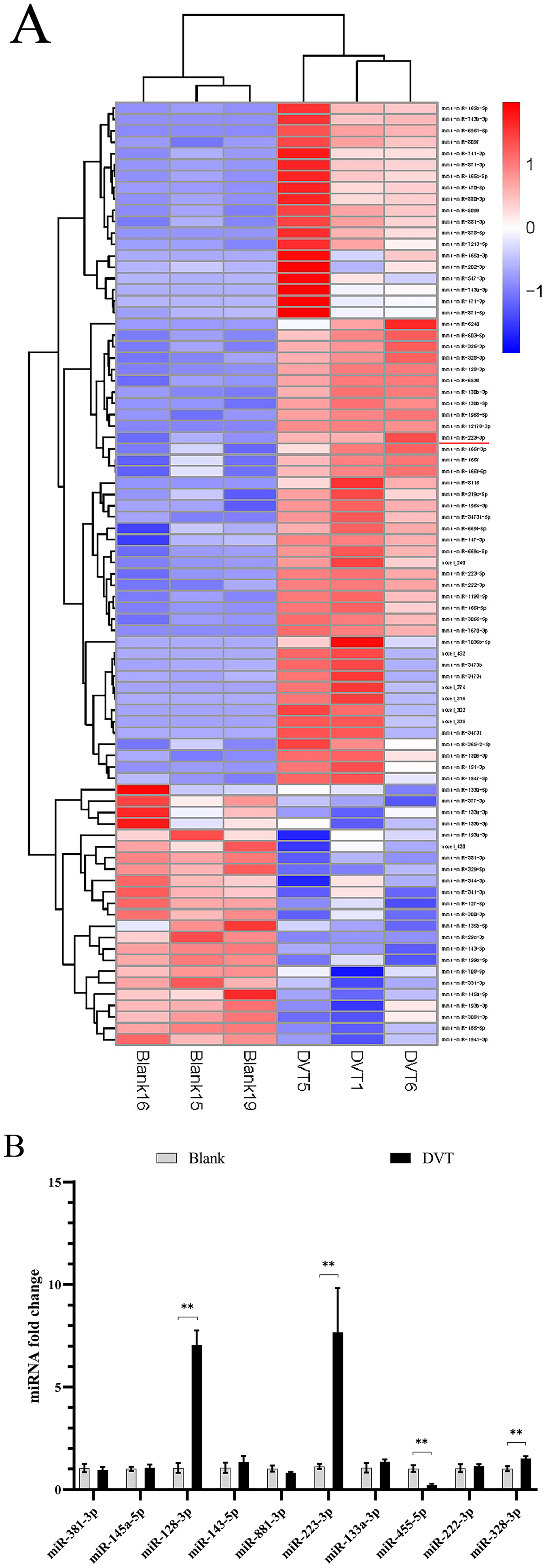
Differential miRNA expression in DVT mice. A. Differential miRNA clustering map. B. The expression of candidate miRNAs in the DVT group compared with those in the blank group. *P < 0.05, **P < 0.01.
To better understand the mechanistic role of miR-223-3p in DVT, we aimed to identify its downstream targets. Previous studies have highlighted the NLRP3 inflammasome as a critical regulator in various inflammatory and vascular disorders, including its potential involvement in DVT. Given the importance of NLRP3 in inflammation and its known role in vascular dysfunction, we hypothesized that NLRP3 could be a key target of miR-223-3p in DVT. This led us to further investigate the interaction between miR-223-3p and NLRP3 as a potential pathway through which miR-223-3p affects DVT.
NLRP3 is a downstream target of miR-223-3p
To identify the downstream target of miR-223-3p in mice with DVT, target genes were predicted and compared with our previous mRNA-seq results (Fig. 2A), which ultimately revealed two promising candidate genes, Nlrp3 and Cers6. The previous mRNA-seq dataset 16 was analyzed via Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis to identify relevant biological functions, and ultimately, 40 pathways were identified between the DVT group and the blank group. The results revealed significant enrichment in the NOD-like receptor signaling pathway, which included Nlrp3 (Fig. 2B). Nlrp3 was found to be a direct target gene, with predicted binding sites for miR-223-3p in both humans and mice (Fig. 2C). Previous studies using dual-luciferase reporter assays confirmed that miR-223-3p can bind to and significantly reduce NLRP3 expression in HUVECs. 13 Therefore, based on these findings, we explored the specific functions and regulatory mechanisms of miR-223-3p in DVT formation. qPCR revealed that Nlrp3 mRNA expression was robustly increased in the vein walls of mice with DVT (P = 0.0385) (Fig. 2D).
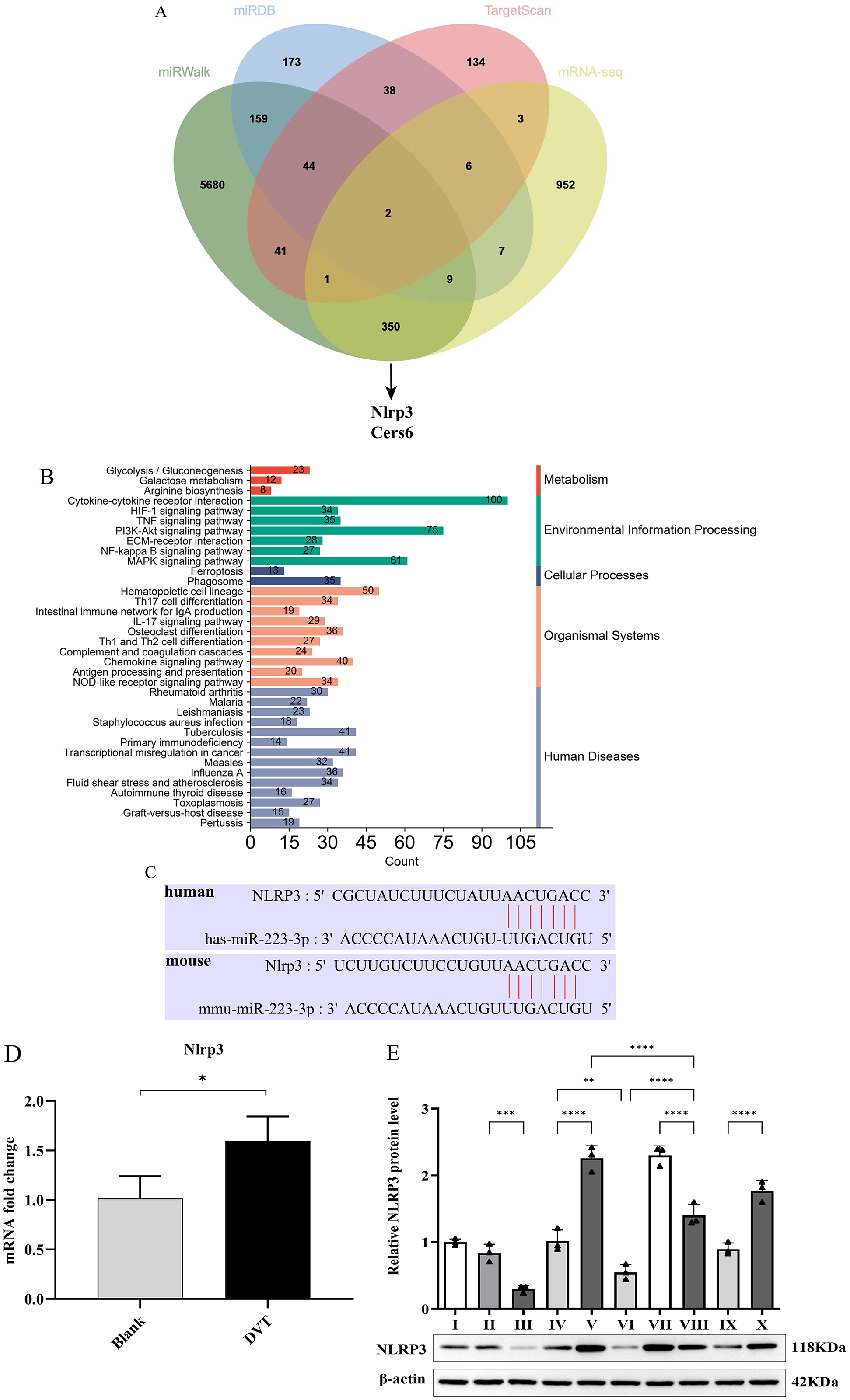
NLRP3 is a downstream target of mir-223-3p. A. Venn diagram showing the predicted target genes of miR-223-3p based on TargetScan, miRDB, miRWalk and mRNA-Seq data. B. Bar diagram of the KEGG pathway enrichment analysis of the mRNA-Seq results. C. The binding sites of miR-223-3p and NLRP3. D. The expression of NLRP3 in the DVT group compared with that in the blank group. *P < 0.05. E. Western blot analysis of NLRP3 in vitro. I: the normal cell group; II: the miR-223-3p-mimic-NC group; III: the miR-223-3p-mimic group; IV: the empty vector control group (transfected with the plasmid PGMLV-CMV-MCS-EF1-ZsGreen1-T2A-Puro); V: the NLRP3 overexpression group; VI: the miR-223-3p-mimic and empty vector control groups; VII: the miR-223-3p-mimic NC and NLRP3 overexpression groups; VIII: the miR-223-3p-mimic and NLRP3 overexpression groups; IX: the miR-223-3p-inhibitor-NC group; and X: the miR-223-3p-inhibitor group. n = 3. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We altered the expression of miR-223-3p in LPS-induced HUVECs using mimics and inhibitors and examined the protein expression of NLRP3. Western blot analysis revealed that NLRP3 expression was lower in HUVECs transfected with the miR-223-3p mimic than in those transfected with the miR-223-3p NC mimic (P = 0.0025). In contrast, the miR-223-3p inhibitor led to the opposite results, and NLRP3 expression was significantly upregulated in the group transfected with the miR-223-3p NC inhibitor (Fig. 2E), which indicates that miR-223-3p inhibits NLRP3 protein expression.
MiR-223-3p suppresses inflammasome activation and vascular dysfunction
To further investigate the potential mechanism by which miR-223-3p reduces thrombus formation, we selected several inflammatory cytokines, which are critical indicators of endothelial inflammation, for further validation in HUVECs and analyzed changes in their expression. These findings helped elucidate the role of miR-223-3p in protecting endothelial cell function and regulating inflammatory responses.
The results indicated that IL-6, IL-1beta, and ICAM-1 expression levels were significantly lower in the miR-223-3p-mimic group than in the miR-223-3p-mimic-NC group and were notably upregulated in the miR-223-3p-inhibitor group compared with those in the miR-223-3p-inhibitor-NC group (P < 0.05) (Fig. 3A-3C). Additionally, the expression of IL-6, IL-1beta, and ICAM-1 in the miR-223-3p mimic and NLRP3 overexpression groups was higher than that in the miR-223-3p mimic and empty vector control groups, which indicates that these suppressive effects were significantly reversed by NLRP3 overexpression. These data suggest that the miR-223-3p-mediated suppression of LPS-induced inflammasome activation in HUVECs depends heavily on NLRP3 and that miR-223-3p is involved in vascular dysfunction by targeting NLRP3.

miR-223-3p suppresses inflammasome activation and vascular dysfunction via the regulation of NLRP3. A-C. Western blot analysis of IL-6, IL-1beta and ICAM-1 levels in vitro. I: the normal cell group; II: the miR-223-3p-mimic-NC group; III: the miR-223-3p-mimic group; IV: the empty vector control group (transfected with the plasmid PGMLV-CMV-MCS-EF1-ZsGreen1-T2A-Puro); V: the NLRP3 overexpression group; VI: the miR-223-3p-mimic and empty vector control group; VII: the miR-223-3p-mimic NC and NLRP3 overexpression group; VIII: the miR-223-3p-mimic and NLRP3 overexpression group; IX: the miR-223-3p-inhibitor-NC group; and X: the miR-223-3p-inhibitor group. n = 3. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Overexpression of miR-223-3p enhances cell viability and promotes HUVEC migration
Upon clearly identifying the regulatory role of miR-223-3p on NLRP3, we further investigated the effects of miR-223-3p on vascular dysfunction, including cell viability and migration ability. The results of the ELISAs of the cell culture supernatant revealed that the levels of IL-6, IL-1beta and TNF-α were lower in the miR-223-3p-mimic group than in the miR-223-3p-mimic-NC group (P < 0.05). Moreover, compared with the empty vector control, the transfection of the NLRP3 plasmid led to the upregulation of IL-6, IL-1beta and TNF-α, whereas these increases in expression levels were significantly reversed by miR-223-3p overexpression (Fig. 4A-C). Next, the viability of HUVECs was tested via CCK-8 assays, and the results revealed that cell viability was strikingly greater in the miR-223-3p-overexpressing group than in the negative control group (Fig. 4D), whereas the effects of miR-223-3p overexpression on cell viability were reversed by NLRP3 upregulation, which suggests that the overexpression of miR-223-3p may contribute to cell proliferation and viability and inhibit inflammation by regulating NLRP3. Finally, the migration ability of HUVECs was tested via migration assays. Compared with the negative control, miR-223-3p upregulation promoted HUVEC migration (P = 0.0019), whereas these effects were reversed by NLRP3 upregulation (Fig. 4E-F). These findings indicate that miR-223-3p facilitates cell migration, which is associated with a decrease in inflammatory cytokines and tissue repair.

Overexpression of miR-223-3p enhances cell viability and promotes HUVEC migration. A-C. The protein expression levels of IL-6, IL-1beta and TNF-α in the culture supernatant of HUVECs were determined by ELISAs. D. The viability of vascular endothelial cells in each group was determined via a CCK-8 assay. E. Transwell assay results showing cell migration and the ratio of the migration area to the initial area after transfection in each group. F. A scratch wound assay was conducted to evaluate the cell migration capacity. Representative images from six experimental groups are shown at 0 h (baseline) and 24 h (post-scratching). For quantification, three images per time point per group were randomly selected for analysis, while only one representative image per condition is displayed. I: The normal cell group; II: the miR-223-3p-mimic-NC group; III: the miR-223-3p-mimic group; IV: the empty vector control group (transfected with the plasmid PGMLV-CMV-MCS-EF1-ZsGreen1-T2A-Puro); V: the NLRP3 overexpression group; VIII: the miR-223-3p-mimic and NLRP3 overexpression group. n = 3. *P < 0.05, **P < 0.01, ***P < 0.001.
Overexpression of miR-223-3p inhibits thrombosis in DVT mice
Based on the in vitro findings that miR-223-3p suppresses inflammation and promotes endothelial cell viability and migration, we sought to evaluate its effects in vivo. Specifically, we investigated whether miR-223-3p could similarly inhibit thrombus formation and reduce vascular inflammation in a mouse model of deep vein thrombosis (DVT). To assess the role of miR-223-3p in vivo, we used a mouse model of DVT, the results of which revealed that the inferior vena cava (IVC) walls were unobstructed in the control group; however, compared with those in the control group, the walls in the DVT group exhibited visible thrombus formation (Fig. 5A), and the wet weights of the thrombi and the weight–to-length ratios were significantly greater in the DVT group. These findings confirmed the successful establishment of the mouse model of DVT. To further investigate the role of miR-223-3p in DVT, mice were injected with agomiR-223-3p before IVC ligation. To validate the effect of miR-223-3p injection, western blotting was used to detect the protein expression of its target gene Nlrp3. As expected, agomiR-223-3p treatment led to a reduction in Nlrp3 levels compared with those in the DVT-NC (P = 0.0014) and agomir-NC groups (P = 0.0036) (Fig. 5B). The weight and weight–to-length ratio of the thrombus are fundamental indices for quantifying the extent of DVT formation. Compared with that in the DVT-NC and DVT-agomir-NC groups, the thrombus weight was significantly lower in the agomiR-223-3p group (P < 0.05) (Figs. 5C and 5D). The qPCR results revealed that, compared with agomir-NC, agomiR-223-3p decreased Il-6 and IL-1beta levels (Fig. 5E). These data suggest that increased miR-223-3p expression can reduce the thrombus burden by alleviating vascular endothelial inflammation. Additionally, the expression of Icam-1 was significantly reduced in the vessel walls of mice in the DVT agomiR-223-3p group. These results indicate that the overexpression of miR-223-3p can inhibit thrombus formation.

Role of mir-223-3p in DVT formation in vivo. A. Images of DVT in mice. B. The expression levels of NLRP3 in the vein walls of the mice, as determined by WB. C and D. The thrombus weight and thrombus weight/length were measured in the different treatment groups. E The expression levels of Il-6, IL-1beta and Icam-1 in the vein walls of the mice, as determined by qPCR. n = 3. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
In this study, we offer new insights into the mechanism of the miR-223-3p-Nlrp3 axis in DVT through systematic experimental validation and data analysis. miR-223-3p has been shown to regulate gene expression by binding to the 3′UTR of target genes, and although previous studies have shown that miR-223-3p is significantly upregulated in the blood of DVT patients, 7 its exact role in DVT pathogenesis has remained elusive. While prior studies have focused mainly on the involvement of miR-223-3p in arterial inflammation,14,15 our research focused on its regulation of venous inflammation, specifically in DVT, which has not been systematically explored previously.
Our bioinformatics analyses suggested Nlrp3 as a potential target of miR-223-3p. To systematically identify possible miR-223-3p target genes, we integrated three independent miRNA prediction databases (TargetScan, miRDB, and miRWalk) with our previously published mRNA-seq dataset from a DVT mouse model. 16 This approach identified Nlrp3 and Cers6 as overlapping candidate genes. However, while Cers6 has not been directly implicated in thrombosis-related pathways, Nlrp3 has been extensively studied in thromboinflammation and hypoxia-induced thrombosis. 11 These findings provided further evidence supporting Nlrp3 as a key downstream target of miR-223-3p for experimental validation. Importantly, the inclusion of mRNA-seq data in our screening pipeline enabled us to refine the list of potential targets by prioritizing genes that were not only predicted by miRNA databases but also differentially expressed in the DVT mouse model. This approach helped exclude less relevant candidates, ensuring that our downstream validation focused on functionally significant targets.
Previous studies using dual-luciferase reporter gene assays have confirmed that miR-223-3p can target NLRP3 and may inhibit inflammasome activation and pyroptosis in HUVECs. 13 Additionally, miR-223-3p has been shown to modulate the expression of IL-6 and IL-1β in THP-1 cells, adipose stem cells, and HUVECs, potentially through NLRP3 regulation.13,15,22 Our study adds to this body of work by suggesting that the overexpression of miR-223-3p may contribute to the downregulation of Nlrp3, IL-1beta, and Il-6 in vivo, which could be associated with a reduced thrombus burden and endothelial inflammation in a DVT mouse model.
Furthermore, we considered other potential targets of miR-223-3p, such as STAT3
23
and IL-6, both of which have been implicated in vascular inflammation and thrombogenesis. STAT3 plays roles in endothelial activation
24
and platelet function,
25
whereas IL-6 is a well-known inflammatory cytokine involved in DVT formation.
20
However, our experimental results suggested that miR-223-3p overexpression did not significantly alter STAT3 protein levels (Supplementary File 1), and the regulation of IL-6 expression by miR-223-3p was not confirmed through dual-luciferase assays. Based on these findings, we focused on NLRP3 as the primary target in this st
Previous studies have demonstrated the potential anti-inflammatory and antithrombotic effects of miR-223-3p in different disease models. For example, in the TAO model, miR-223-3p has been reported to reduce thrombosis and local inflammation by downregulating NLRP3. 14 However, TAO is characterized by chronic arteritis, whereas sterile inflammation in DVT is triggered mainly by thrombosis, representing a fundamental difference in the associated inflammatory mechanisms. Sterile inflammation in the vessel wall can contribute to thrombosis development, even if the vessel integrity is not disrupted. 26 In DVT, the canonical Nlrp3 inflammasome participates in NETosis, and Nlrp3 deficiency has been shown to reduce the density of neutrophil extracellular traps (NETs) in thrombi. 27 Additionally, in a gout model, miR-223-3p was found to suppress inflammation induced by monosodium urate crystals via NLRP3 regulation. 28 Although these studies support a regulatory role for miR-223-3p in inflammation, our study provides evidence supporting its potential function in endothelial inflammation and thrombosis in DVT.
This study suggests a potential mechanism by which miR-223-3p may regulate the NLRP3 inflammasome in venous environments, specifically by modulating endothelial inflammation and thrombosis formation, a function that has been less extensively studied.
In addition to these findings, miR-223-3p has been implicated in broader vascular dysfunction beyond DVT, with increased miR-223-3p levels in plasma and atherosclerotic plaques associated with reduced inflammation and lesion size in subjects with atherosclerosis.29,30 miR-223-3p has also been suggested to reduce high glucose-induced endothelial cell injury by targeting NLRP3, decreasing apoptosis, and promoting cell migration. 31 Although previous studies have explored the role of miR-223-3p in endothelial cells, particularly in arterial diseases such as atherosclerosis, our study provides evidence supporting the idea that miR-223-3p may increase HUVEC viability and migration by downregulating NLRP3, an effect that was partially reversed by NLRP3 overexpression. These findings suggest a potential role for miR-223-3p in endothelial biology during DVT.
Despite the significance of our findings, some limitations should be considered. Our results are based on a mouse model, and human samples would be valuable for further validation. Additionally, although we focused on the potential role of miR-223-3p in regulating NLRP3 in endothelial cells, further research could explore its effects on other cell types, such as smooth muscle cells and platelets, in the context of thrombus formation. Moreover, while miR-223-3p shows complete sequence conservation between mice and humans and NLRP3 is moderately conserved as a target, potential species-specific differences in downstream pathways should be investigated to ensure the translational relevance of our findings. In the DVT group, the upregulation of miR-223-3p may represent a compensatory response to inflammation rather than a complete suppressive mechanism on NLRP3. However, strong proinflammatory signaling, particularly NF-κB activation, 32 has been shown to promote NLRP3 transcription independently of miR-223-3p regulation, potentially limiting its inhibitory effect. Additionally, competing endogenous RNAs and RNA-binding proteins may sequester miR-223-3p, further modulating its regulatory efficiency.33–35 This incomplete suppression of NLRP3 by endogenous miR-223-3p indicates that its regulatory effect may be dose dependent, as suggested by the stronger inhibition observed following exogenous miR-223-3p mimic administration. These findings highlight the complex interplay between miRNAs and inflammation, supporting the need for further investigations into the mechanisms that regulate miRNA activity under thromboinflammatory conditions.
miR-223-3p may also regulate other targets and pathways involved in DVT, suggesting the need for a broader exploration of its function. Future studies could explore the broader regulatory network involving miR-223-3p in DVT and its potential therapeutic applications. Although the role of miR-223-3p in regulating inflammation has been reported previously, our study provides insights into its possible function in venous thromboinflammation.
Unlike prior studies on arterial diseases or systemic inflammation, our findings suggest that miR-223-3p may modulate thrombus formation in a DVT model by targeting NLRP3. However, the upregulation of miR-223-3p in DVT does not fully suppress NLRP3, indicating additional regulatory mechanisms. Persistent NLRP3 expression may result from ceRNA sequestration or inflammatory pathway activation (e.g. NF-κB). The stronger inhibition observed with the miR-223-3p mimic highlights its dose-dependent effect. Future studies may investigate these interactions to better understand miRNA functions in thromboinflammation.
Although formal sample size calculations were not conducted in this study, the observed significant differences between groups and the effect sizes suggest the biological relevance of our findings. Nonetheless, we recognize that a more rigorous approach to sample size calculation and power analysis would further strengthen the reliability of the results in future studies.
Conclusions
Our study suggests that miR-223-3p may act as a regulator of thromboinflammation in deep vein thrombosis. By downregulating Nlrp3, miR-223-3p appears to reduce the thrombus burden and inflammatory cytokine levels. AgomiR-223-3p treatment was associated with a decrease in thrombosis and an improvement in endothelial function, indicating its potential as a therapeutic target. In vitro, miR-223-3p may inhibit NLRP3-mediated inflammation and increase endothelial cell viability and migration. These findings highlight a possible role for miR-223-3p in venous thrombosis and warrant further investigation to explore its therapeutic applicability.
Supplemental Material
sj-docx-1-sci-10.1177_00368504251337526 - Supplemental material for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model
Supplemental material, sj-docx-1-sci-10.1177_00368504251337526 for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model by Ji Luo, Zheng Lei, Hongyu Zheng and Rudan Zhou in Science Progress
Supplemental Material
sj-pdf-2-sci-10.1177_00368504251337526 - Supplemental material for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model
Supplemental material, sj-pdf-2-sci-10.1177_00368504251337526 for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model by Ji Luo, Zheng Lei, Hongyu Zheng and Rudan Zhou in Science Progress
Supplemental Material
sj-docx-3-sci-10.1177_00368504251337526 - Supplemental material for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model
Supplemental material, sj-docx-3-sci-10.1177_00368504251337526 for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model by Ji Luo, Zheng Lei, Hongyu Zheng and Rudan Zhou in Science Progress
Supplemental Material
sj-docx-4-sci-10.1177_00368504251337526 - Supplemental material for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model
Supplemental material, sj-docx-4-sci-10.1177_00368504251337526 for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model by Ji Luo, Zheng Lei, Hongyu Zheng and Rudan Zhou in Science Progress
Supplemental Material
sj-docx-5-sci-10.1177_00368504251337526 - Supplemental material for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model
Supplemental material, sj-docx-5-sci-10.1177_00368504251337526 for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model by Ji Luo, Zheng Lei, Hongyu Zheng and Rudan Zhou in Science Progress
Supplemental Material
sj-docx-6-sci-10.1177_00368504251337526 - Supplemental material for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model
Supplemental material, sj-docx-6-sci-10.1177_00368504251337526 for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model by Ji Luo, Zheng Lei, Hongyu Zheng and Rudan Zhou in Science Progress
Supplemental Material
sj-docx-7-sci-10.1177_00368504251337526 - Supplemental material for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model
Supplemental material, sj-docx-7-sci-10.1177_00368504251337526 for A novel role of miR-223-3p in reducing NLRP3-mediated inflammation and deep vein thrombosis in a mouse model by Ji Luo, Zheng Lei, Hongyu Zheng and Rudan Zhou in Science Progress
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Major Science and Technology Project of Yunnan Provincial Department of Science and Technology, Yunnan Provincial Orthopedic and Sports Rehabilitation Clinical Medicine Research Center, The General Project of Yunnan Basic Research Program, the Joint Project of Chengdu Medical College and The First People's Hospital of Ziyang (grant number 202102AA310068, 202301AT070104, 2022LHZY08).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.