
Abstract
Benign recurrent intrahepatic cholestasis (BRIC) is a rare genetic liver disorder characterized by recurrent episodes of jaundice and severe pruritus without significant liver damage. Here, we present the case of a man in his early 20s, standing 143 cm tall, who has experienced recurrent jaundice and pruritus over an 18-year period. Laboratory investigations and liver biopsy examination indicated typical intrahepatic cholestasis with normal gamma-glutamyl transpeptidase levels, excluding viral, metabolic, and autoimmune causes. Further genetic analysis confirmed the diagnosis of BRIC with two compound heterozygous mutations in ATP8B1 gene on chromosome 18: NM_001374385.1: c.2081T > C p. (Ile694Thr) (rs541474497) in exon 18 and NC_000018.10 (NM_001374385.1): c.1631-2A > G in intron 15. To our knowledge, the latter has not been reported. Family lineage analysis and Sanger sequencing showed that these mutations were de novo rather than hereditary. Treatment with ursodeoxycholic acid and cholestyramine resulted in complete resolution of jaundice and pruritus, with normalization of serum bilirubin level after six months of therapy.
Introduction
Benign recurrent intrahepatic cholestasis (BRIC) is a rare genetic disorder characterized by self-limiting recurrent jaundice while accompanying with severe pruritus. 1 Benign recurrent intrahepatic cholestasis episodes can vary widely in duration, lasting from several weeks to months, or even years. 2 While the precise epidemiology of BRIC remains unclear, its global incidence is estimated at approximately 1 in 50,000–100,000 individuals. 3 Benign recurrent intrahepatic cholestasis can occur at any age, but in most cases, the initial symptoms arise in early adolescence. During episodes, the levels of gamma-glutamyl transpeptidase (GGT) remain normal or slightly elevated, but serum bilirubin and alkaline phosphatase (ALP) levels are significantly increased. 4 Benign recurrent intrahepatic cholestasis episodes may emerge spontaneously or be triggered by factors such as infection, pregnancy, oral contraceptives, or stress.2,5,6 Benign recurrent intrahepatic cholestasis has two subtypes including BRIC type 1 (BRIC-1) and BRIC type 2 (BRIC-2). Both subtypes are marked by a gradual return to normal clinical manifestations and blood biochemical indicators, without progression to cirrhosis, liver failure, or growth impairment. 7 Here, we report the case of a male patient with growth retardation due to BRIC. Genetic analysis indicated that two spontaneous compound heterozygous mutations in the ATP8B1 gene are associated with recurrent intrahepatic cholestasis, suggesting the importance of genetic screening, timely diagnosis, and therapy for this rare genetic disorder in clinical practice.
Case presentation
A male patient in his early 20s presented to the Internal Medicine Department at Jiading District Central Hospital, Jiading District, Shanghai, in March, 2024, with complaining of jaundice, arthralgia, and diffuse pruritus persisting for approximately 18 years. He had no fever, chills, skin rash, history of drug use, or family history of liver disease. These symptoms, accompanied by long-term intermittent diarrhea, had first manifested when he was two years old. His appetite was normal without vomiting, which means adequate nutritional status. There was no history of similar conditions among his nonconsanguineal parents and elder brother. The patient's mother reported that her son first developed generalized jaundice and pruritus in March 2006, at age 2, and was subsequently admitted to a local hospital, where the laboratory findings revealed marked elevation of serum bilirubin and ALP levels, with normal liver function and GGT. Other detailed laboratory test results are shown in Table 1. Abdominal computed tomography indicated mild hepatosplenomegaly, leading to a diagnosis of cholestasis syndrome. He was prescribed with ursodeoxycholic acid (UDCA), at the dose of 0.5 g/d. His condition improved gradually, and liver function tests normalized after about 2 months. The patient experienced two subsequent hospitalizations following UDCA discontinuation: in August 2015 (at the age of 11) and August 2020 (at the age of 16.5). Neither episode was preceded by infection, drug use, or vaccination. Laboratory workups during these hospitalizations were consistent with his first presentation. In both instances, symptoms resolved within about 2 months of resuming UDCA treatment. During asymptomatic periods, both clinical symptoms and laboratory parameters remained within normal ranges. Based on his clinical manifestations, liver biopsy, and gene analysis were recommended but refused by his parents due to their financial constraints.
Laboratory data of the patient during the past hospitalizations.
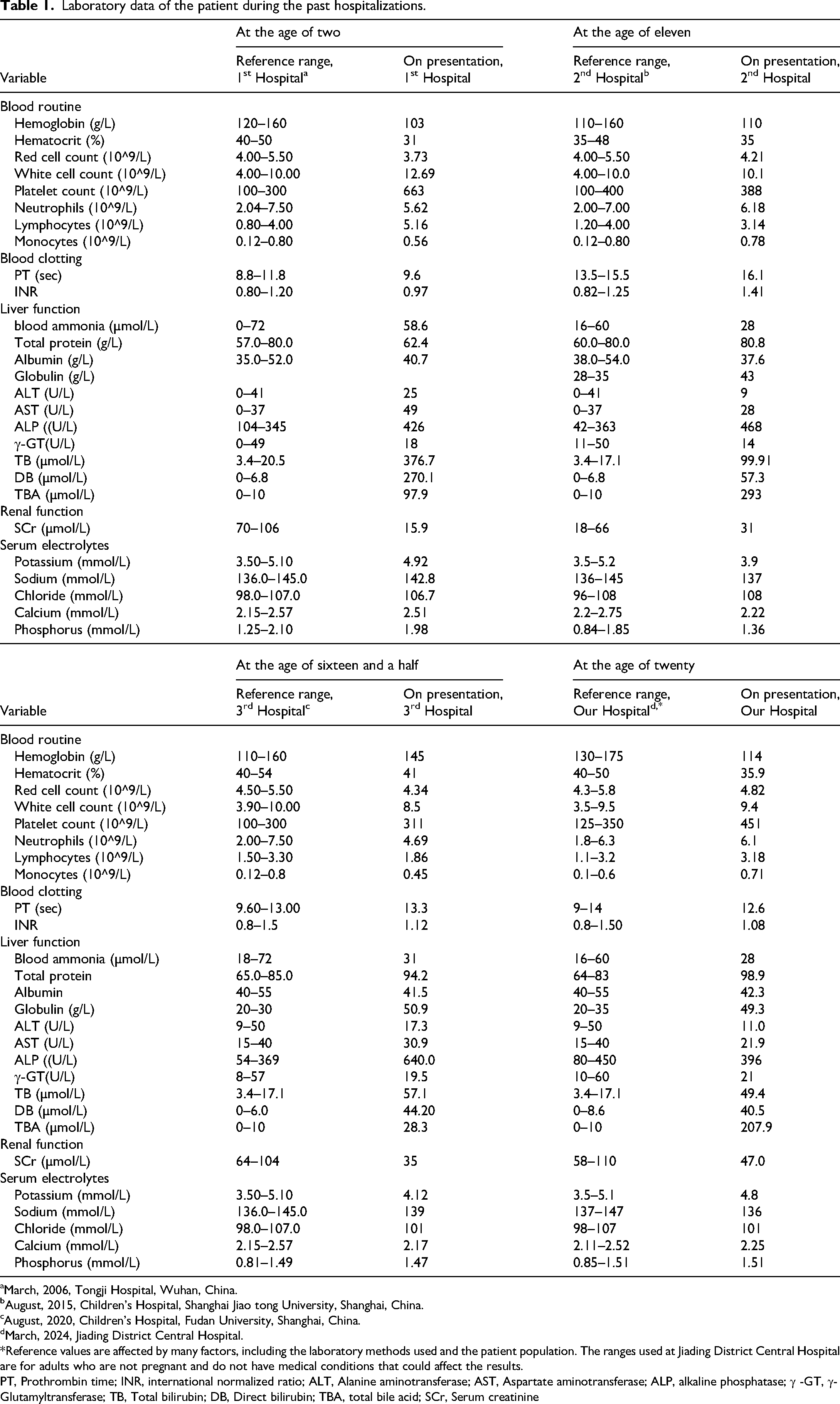
aMarch, 2006, Tongji Hospital, Wuhan, China.
bAugust, 2015, Children's Hospital, Shanghai Jiao tong University, Shanghai, China.
cAugust, 2020, Children's Hospital, Fudan University, Shanghai, China.
dMarch, 2024, Jiading District Central Hospital.
*Reference values are affected by many factors, including the laboratory methods used and the patient population. The ranges used at Jiading District Central Hospital are for adults who are not pregnant and do not have medical conditions that could affect the results.
PT, Prothrombin time; INR, international normalized ratio; ALT, Alanine aminotransferase; AST, Aspartate aminotransferase; ALP, alkaline phosphatase; γ -GT, γ-Glutamyltransferase; TB, Total bilirubin; DB, Direct bilirubin; TBA, total bile acid; SCr, Serum creatinine
At the current admission to our hospital, physical examination disclosed a height of 143 cm and body weight of 39 kg, with severe xanthochromia of skin and sclera (Figure 1(a) and (b)). The body builds of his parents and elder brother were within normal ranges (Figure A in Supplemental Materials). His abdomen was soft and nontender, with palpable splenomegaly but no jugular vein distention or ascites. No spider angiomas or palmar erythema were observed on his skin. His blood ammonia level was normal, with no signs of involuntary movements or cognitive difficulties indicating central nervous system involvement or hepatic encephalopathy. His uranalysis was positive for bilirubin. Kayser–Fleischer rings were not found in either eye by slit-lamp examination.

(a) and (b) The patient's body height of 143 cm, and yellow skin and sclera, respectively; (c) liver histology showed lobular structure remains intact (hematoxylin and eosin stain × 100), (d) bile plugs, mild hepatic plate swelling, diffuse hydropic degeneration of hepatocytes, and minor fibrous connective tissue were also observed (hematoxylin and eosin stain × 400).
Virologic studies were negative for hepatitis A, B, C, D, and E viruses, as well as cytomegalovirus, Epstein–Barr virus, herpes simplex virus, and rubella. Autoimmune markers (antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, anti-liver–kidney microsomal antibodies) and serum ceruloplasmin levels were all within normal ranges. Serum 25-(OH)-Vitamin D was 6.80 ng per liter (a level below 20 means deficiency). The serum levels of parathormone (PTH), insulin-like growth factor-1 (IGF-1), and thyroid-stimulating hormone (TSH) were 42.3 pg/mL (reference range, 15–68.3), 82.00 ng/mL (reference range for 16–20-year-old adult, 75–850), and 5.07 μIU/mL (reference range, 0.35–4.94), respectively. Serum ceruloplasmin, 24-h urinary copper and calcium levels were 56.03 mg/dL (reference range, 15–30), 23 µg/24 h (reference range, 15–30), and 1.1 mmol/24 h (reference range, 2.5–7.5), respectively. His cortisol rhythm, serum thyroid and growth hormone levels, and tumor biomarkers were unremarkable. The remaining laboratory test results are shown in Table 1. Abdominal magnetic resonance imaging revealed splenomegaly, while magnetic resonance cholangiopancreatography showed mild gallbladder enlargement and cyst-like dilatation of the main pancreatic duct. Radiography of the left wrist indicated a bone age of 17-year-old (Figure B, C, D in Supplemental Materials). Bone densitometry demonstrated severe osteoporosis, with a Z score of −5.0 in lumbar vertebrae. With patient consent, a liver biopsy was performed, revealing intact lobular structure with few lymphocytes and neutrophils in hepatic lobules and portal areas, along with bile plugs and slight fibrous connective tissue proliferation (Figure 1(c) and (d)). Immunohistochemical staining of bile salt export pump (BSEP) and multidrug-resistance protein 3 (MDR3) were unremarkable.
Whole exome sequencing and third-generation sequencing of targeted fragment
1. Sample preparation
Whole blood samples were collected from the proband, his biological brother, and parents for extracting total genomic DNA (gDNA). After collection, total gDNA was extracted within 2 h using the blood sample DNA isolation kit (Qiagen Co., Ltd Germany).
2. Whole exome sequencing
DNA libraries were prepared and exome capture was performed using the Twist Human Core Exome Kit (Twist Bioscience), which targets approximately 35.8 Mb of coding regions across the human genome. The enriched libraries were pooled in equimolar ratios and sequenced on BGI T7 platform (BGI, China) using 150 bp paired-end reads. Sequencing depth was targeted at a minimum of 100× coverage for each sample to ensure high-confidence variant calling. Sequencing data were aligned to the human reference genome (hg19) using the Burrows–Wheeler Aligner (BWA-MEM, v0.7.17). Duplicate reads were marked and removed using Picard Tools (v2.23.8). Variant calling was performed using the Genome Analysis Toolkit (GATK, v4.2.0) following best practices, including base quality score recalibration and variant filtration. Annotation of variants was conducted using ANNOVAR, and functional impact was predicted using databases such as ClinVar, dbSNP, and gnomAD. Candidate variants were validated using Sanger sequencing on an ABI 3730xl DNA Analyzer (Applied Biosystems, USA).
3. Long-Range polymerase chain reaction (PCR) and Nanopore Sequencing
The PCR amplification of targeted amplicon was performed according to KOD FX Neo (KFX-201, TOYOBO, Japan). The primers for the targeted amplicon in ATP8B1 gene are shown in Table 2. The cycling conditions for the first round for amplification of targeted amplicon in ATP8B1 gene were denaturation at 94 °C for 1 min, 98 °C for 10 s, annealing at 63 °C for 30 s, and extension at 68 °C for 7 min, for 30 cycles; and for the second round, denaturation at 94 °C for 3 min, 98 °C for 10 s, annealing at 60 °C for 30 s, and extension at 68 °C for 7 min, for 5 cycles. The length of the targeted region was 6760 bp. Library was constructed by Ligation Sequencing Kit (SQK-LSK110, Nanopore, UK), and then sequenced by PromethION 2 Solo (Nanopore, UK).
Primers for PCR used in gene amplification of the study specimens.

Familial lineage analysis
Whole exome sequencing for the patient identified two heterozygous mutations: NM_001374385.1: c.2081T > C p. (Ile694Thr) (rs541474497) in exon 18 and NC_000018.10 (NM_001374385.1): c.1631-2A > G in intron 15, the gene encoding the FIC1 protein. Single nucleotide polymorphism–based kinship analysis confirmed first-degree biological relationships between the proband and family members (relatedness Phi of proband vs. mother, father, and brother are 0.2295, 0.2266, and 0.2277, respectively; reference range: 0.157–0.354), with no consanguinity between parents (Phi 0.024; reference range <0.0442). Whole exome sequencing of biological parents and elder brother showed no mutations at these sites. Long-range PCR amplification followed by Oxford Nanopore sequencing analysis demonstrated that the two variants were in trans (Figure E in Supplemental Materials), indicating they are located on separate alleles. According to ACMG guideline, both variants were classified as pathogenic, c.2081T > C p. (Ile694Thr) (PS2 + PM2_ Supporting + PM3 + PM5 + PP2 + PP3_Strong + PP4), and c.1631-2A > G (PVS1 + PS2 + PM2_Supporting + PM3 + PP4) (Genetic Test Report in Supplemental materials). The raw sequence data have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA010767) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human.
Diagnosis, therapeutic intervention, follow-up, and outcomes
These findings supported a diagnosis of BRIC, along with growth impairment, and osteoporosis. Following treatment consent, the patient was started with UDCA at 1 g/day, along with cholestyramine at 4 g daily, and calcium supplementation, at a dose of 0.3 g, three times daily, for osteoporosis. At three months follow-up, where substantial improvements were noted in jaundice, arthralgia and generalized pruritus, with normalization of liver function and serum bilirubin. He was advised to have follow-up routinely every 6 months.
The reporting of this study conforms to CARE guidelines. 8 Written informed consent was obtained from the patient and his family members prior to the study initiation and for the publication of their clinical information.
Discussion
Benign recurrent intrahepatic cholestasis is very rare in East Asian countries, and the paucity of reports has led to obscurity regarding the clinical, genetic and prognostic features of patients from this region. 7 The clinical features of BRIC are characterized by jaundice, pruritus, marked elevation of serum ALP and bilirubin, alongside normal or mild elevated GGT. 3 Benign recurrent intrahepatic cholestasis can be classified into two types: Type 1, which involves a mutation in the ATPase Phospholipid-Transporting 8B1 (ATP8B1) gene, encoding the hepatocanalicular flippase for phosphatidylserine; and Type 2, which involves a mutation in the ATP-Binding Cassette Subfamily B Member 11 (ABCB11) gene, encoding the hepatocellular BSEP. 9 A recent study from Japan confirmed that a female adult suffered from BRIC due to biallelic MYO5B mutations. 10 Chen et al. reported a novel homozygous variant, including c.1817T>C and p.I606T, detected in a Chinese male patient with BRIC-1 in his ATP8B1 gene, and deduced that this variant was potentially inherited from his parents. 11 Lee et al. presented a seven-year-old boy with BRIC who exhibited growth retardation, with genetic analysis revealing a heterozygote frameshift mutation by insertion of a nucleotide C at 2610_2611 (p.K871Q-fsX). 12 In this study, we reported a growth-impaired young male who underwent four attacks of cholestasis. Whole exome sequencing revealed two heterozygous mutations in ATP8B1. The first variant (c.2081T > C) has a known association with BRIC. 13 While the second variant (c.1631-2A > G) is a novel splicing mutation of uncertain clinical significance. Notably, familial lineage analysis confirmed the absence of both variants in the patient's biological parents and elder brother, a rare finding that suggests these two variants were presumed to be de novo or both parents may harbor germline mosaicism.
Generally, the episodic nature of BRIC indicated that cholestatic symptoms persisted for several weeks to months before spontaneous resolution. Common triggers include viral infections, hormonal disturbances (such as those induced by oral contraceptive pills or pregnancy), and less commonly, certain medications and other factors. 14 However, our patient's course did not align with these typical triggers. Instead, the pattern of symptom recurrence following UDCA discontinuation suggests that abrupt cessation of UDCA therapy may serve as an additional trigger for disease episodes.
Previous studies confirmed BRIC is associated with a favorable prognosis and does not usually lead to cirrhosis or liver failure. In contrast, progressive familial intrahepatic cholestasis (PFIC), another autosomal recessive disorder, characterized by persistent cholestasis with a strong phenotype and poor prognosis, often progresses to end-stage liver disease.4,7,14 Nevertheless, there have been reports of progression from BRIC to PFIC, 4 suggesting the possible nature of both diseases as being part of the same spectrum. A multicenter cohort study found growth failure with stunting is more predominant in PFIC1 disease compared to BSEP or MDR3 disease, 15 as observed in our patient's clinical presentation. Addressing this overlapping phenomenon requires further clinical and molecular investigations to elucidate the underlying mechanisms and to determine the most appropriate classification. Understanding the progression from BRIC to PFIC could have significant implications for patient's management and prognosis.
Majority of patients with BRIC exhibit normal growth and development with satisfactory long-term outcomes. 7 Nagasaka et al. reported two patients with familial intrahepatic cholestasis, caused by ATP8B1 mutations who had episodic hypocalcemia as a result of pseudohypoparathyroidism (PHP), suffered from growth impairment and osteoporosis. They proposed effects of excessive urinary calcium resulting from PHP during cholestatic attacks together with long-standing increased bone resorption activity between attacks might result in accelerated bone loss and growth impairment in these patients.16,17 In contrast, our patient did not exhibit the typical biochemical markers associated with PHP. Normal serum levels of calcium and phosphorus were maintained during four hospitalizations, and PTH, IGF-1, and TSH were decreased at the current hospital stay combined with decreased 24-h urinary calcium, thus excluding the diagnoses of PHP. The underlying pathogenesis may be attributed to the role of the ATP8B1 gene, which encodes an aminophospholipid flippase anchored to the canalicular membranes of cholangiocytes and hepatocytes, pivotal for normal bile flows across membranes—likely by maintaining an asymmetric distribution of different phospholipids between the inner and outer leaflets of the plasma membrane. 18 ATP8B1 deficiency may lead to instability of membrane, lack of cholesterol in the apical membrane, and dysfunction of transmembrane transporters such as the BSEP, the key transporter of bile salts into the canaliculus. Thus, it is unsurprising that such malfunction could lead to cholestasis, reduced digestion and absorption of lipids, and vitamin D deficiency. ATP8B1 is also expressed in extrahepatic tissues, such as small intestine and pancreas, which may explain the diarrhea observed in BRIC-1 patients. 5 Additionally, repeated occurrence of hyperbilirubinemia may bring damage to the proliferative capacity of osteocytes, a condition that can be reversed with UDCA and cholestyramine administration.19,20 Further investigation and accumulation of additional case reports are crucial to advancing our understanding of the pathophysiology of BRIC and its potential long-term complications.
Conclusion
We reported a young man with BRIC-1 who also displayed growth retardation and osteoporosis. Comparative genetic analysis of familial lineage identified two functional mutations in the ATP8B1 gene. This study highlights the importance of recognizing clinical symptoms and using effective diagnostic and therapeutic approaches for BRIC-1.
Patient perspective
Living with BRIC-1 has been a lifelong challenge. Since childhood, I’ve dealt with episodes of jaundice and severe itching that came and went without warning. These symptoms made me feel different and frustrated, especially as I grew up smaller than my peers. Getting diagnosed with BRIC was a mix of relief and worry. Finally, I had answers after years of uncertainty, but I also realized this was something I’d have to manage forever. The genetic testing that found mutations in the ATP8B1 gene was a big moment—it helped me understand why my body was acting this way. Starting treatment with UDCA and cholestyramine changed everything. The itching and yellowing of my skin improved, and I began to feel more like myself. Managing osteoporosis with calcium supplements has been another part of the journey, but I’m grateful for the progress I’ve made. The support from my doctors and family has been incredible. My medical team has always been patient and thorough, and my family has been there for me every step of the way. While there's no cure for BRIC, I feel hopeful about the future. With the right treatment and support, I’ve learned to manage my condition and live a full life. Sharing my story is important to me because I want others with BRIC to know they’re not alone.
Supplemental Material
sj-pdf-1-sci-10.1177_00368504251335415 - Supplemental material for A sporadic case of benign recurrent intrahepatic cholestasis in a growth-impaired young male
Supplemental material, sj-pdf-1-sci-10.1177_00368504251335415 for A sporadic case of benign recurrent intrahepatic cholestasis in a growth-impaired young male by Jiasheng Shao, Rong Fan, Minhua Shao, Min Dai, Gang Huang, Qiang Guo and Liou Cao in Science Progress
Footnotes
Acknowledgements
The authors are indebted with the patient and his family involved in the study. The authors also thank Jiayan Liu, Xin Qiao, Guanghui Li for their medical advices and consultations. Finally, the authors thank Shanghai WeHealth BioMedical Technology Co., Ltd, China, for providing sequencing service and familial lineage analysis.
Ethical Considerations
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of Jiading District Central Hospital Affiliated Shanghai University of Medicine & Health Sciences (Ethics approval number: 2023-B-03-03, Date of Approval: May 6, 2024). All information about the patient was anonymized.
Informed consent
Written informed consent was obtained from the patient and his family prior to the study initiation, and we have been authorized for the publication of any potentially identifiable images or data included in this article.
Author contributions/CRediT
Conceptualization and design: Jiasheng Shao and Liou Cao; data collection: Jiasheng Shao and Ron Fan; image data: Minhua Shao, Min Dai, and Gang Huang; writing—original draft preparation: Jiasheng Shao and Rong Fan; funding acquisition: Liou Cao and Qiang Guo. All authors have read and agreed to the published version of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The case study was supported by the Science and Technology Commission of Jiading District (Grant No. JDKW-2024-0039) and Shanghai University of Medicine and Health Sciences Clinical Research Centre for Metabolic Vascular Diseases Project (20MC2020004).
Conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The original contributions presented in the study are included in the article/supplemental material; further inquiries can be directed to the corresponding authors.
Supplemental material
Supplemental material for this article is available online.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.