
Abstract
Objective:
To explore abdominal aortic aneurysm (AAA) pathogenesis and identify early diagnostic markers, providing a theoretical basis for novel preventive and therapeutic strategies.
Methods:
Gene expression profiles were retrieved from the Gene Expression Omnibus database (datasets: GSE7084, GSE47472, and GSE57691) comprising messenger RNA data from the aortic samples of 69 patients with AAA and 25 non-AAA controls. Data were merged and normalized; bioinformatics analysis was conducted on upregulated differentially expressed genes.
Results:
C-X-C motif chemokine ligand 8 (CXCL8) was prominently involved in regulating the chemokine signaling pathway. CXCL8 expression was significantly higher in the aortic walls of patients with AAA than that of controls. NLRP3, interleukin (IL)-18, and IL-1β expression levels were upregulated in patients with AAA and positively correlated with CXCL8 expression. CXCL8 may directly or indirectly interact with NLRP3.
Conclusions:
CXCL8 was upregulated in patients with AAA and induced inflammatory cell infiltration and cytokine secretion. CXCL8-induced NLRP3 inflammasome regulation triggered pyroptosis in vascular smooth muscle cells, exacerbating inflammation and tissue damage in the aortic wall. This degeneration of the aortic media accelerated AAA progression.
Introduction
Abdominal aortic aneurysm (AAA) is a vascular condition characterized by the localized dilation of the abdominal aorta due to aortic wall degradation and the pathological expansion of the lumen. AAA is the most common type of aneurysm, with a mortality rate of 65% to 85% following rupture. 1 AAA occurrence and development involve complex pathophysiological processes characterized by the loss of aortic vascular smooth muscle cells (VSMCs). Although the precise mechanisms underlying AAA are not fully understood, a close link has been reported between AAA onset and progression and inflammatory responses in the aorta.2,3
In the early stages of AAA, lymphocytes, monocytes, macrophages, neutrophils, and other inflammatory cells infiltrate the aortic wall and release inflammatory factors, increasing VSMC death.4,5 Proteases, cell adhesion molecules, and reactive oxygen species are also produced, further damaging the aortic wall.6,7 In most cardiovascular diseases, vascular injury is closely associated with inflammation, leading to smooth muscle cell necrosis and vascular wall structural damage.8,9 Extensive lymphocyte and macrophage infiltration into the aortic media and smooth muscle cell loss suggest that inflammatory cells may migrate from the aortic adventitia to the media and intima.10,11 Hence, inflammatory responses contribute to AAA progression in multiple stages, potentially representing a common pathway influencing AAA pathogenesis.12,13
Continuous advances in RNA sequencing, gene microarrays, and related technologies have improved accuracy and efficiency while reducing costs, leading to large-scale disease-related databases. Utilizing bioinformatics to analyze these databases, researchers can quickly identify genes involved in AAA development and progression.
In this study, bioinformatics analysis was employed to identify and validate diagnostic markers for AAA, elucidate the molecular mechanisms of AAA, and provide a foundation for novel therapeutic and diagnostic strategies.14,15
Methods
Human aorta abdominalis tissues
Abdominal aorta blood and arterial tissue were collected from patients who underwent abdominal aorta artificial vascular replacement for abdominal aneurysm between January 2024 and June 2024 at the Department of Vascular Surgery, Xiamen Branch of Zhongshan Hospital, Fudan University. All patients were diagnosed with abdominal aneurysms, and detailed clinical and pathological data were collected.
Data collection and differential gene expression analysis
Gene expression datasets GSE7084, GSE47472, and GSE57691 were downloaded from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/).16–18 These datasets were submitted by the same research team at different times and were analyzed using the same platform, ensuring consistency in sample sourcing and sequencing. The GSE7084, GSE47472, and GSE57691 datasets contain aortic samples from 69 patients with AAA and 25 non-AAA controls. These datasets were subsequently merged and normalized using the “sva” package in R to remove batch effects. Differentially expressed genes (DEGs) were identified using the “limma” package in R.
The study was approved by the Institutional Review Board of the Xiamen Branch of Zhongshan Hospital, and written informed consent was obtained from all patients. This study was conducted in accordance with the Helsinki Declaration of 1975, revised in 2013.
Gene module functions and pathway enrichment
Gene ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed using the ClusterProfiler package in R 3.6.1 to identify the functional roles of upregulated DEGs. Genes were mapped to KEGG pathways, with a Benjamini-corrected p-value of 0.05 as the threshold for identifying significant GO terms and KEGG pathways. Cytoscape software (http://cytoscape.org/) was employed to generate a protein–protein interaction (PPI) network in which nodes represented proteins and edges represented their interactions.
Identification and exploration of key genes
The STRING database (for PPI networks and functional enrichment analysis; http://string-db.org) was used to construct a PPI network for upregulated DEGs in the AAA and control groups to identify key genes. The PPI network was visualized using Cytoscape software, and molecular complex detection (MCODE) was applied for module analysis. Modules were determined to be critical when they included more than five nodes and the MCODE score exceeded 5.
Reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from cell lines and tissue samples using TRIzol® reagent (Invitrogen, Carlsbad, CA, USA). First-strand cDNA was synthesized with PrimeScript™ RT Master Mix (Takara Biotechnology, Ltd, Dalian, China) according to the manufacturer's protocol. Subsequently, quantitative PCR examined the expression levels of CXCL8 and NLRP3 using SYBR Green PCR Master mix (Takara Biotechnology, Ltd) on a Bio-Rad Real-Time PCR instrument (Bio-Rad Laboratories, Inc., Hercules, CA, USA). GAPDH served as the internal reference gene. The primer sequences were as follows: CXCL8 forward: 5′-TGGCTGGAGTGTTGCAGAGTT-3′, reverse: 5′-TCTGGGGGAAAGCTGTGGA-3′; NLRP3 forward: 5′-ATGCCGTATCTGGTTGTGTT-3′, reverse: 5′-GACTTAGGAAGAGCTGAGCA-3′. GAPDH: Forward primer: 5'-TGCACCACCAACTGCTTAGC-3′ Reverse primer: 5′-GGCATGGACTGTGGTCATGAG-3′. Data were analyzed using the 2−ΔΔCt method.
Immunohistochemistry (IHC)
Tissue samples were fixed using fixatives, such as formaldehyde, dehydrated, cleared, embedded in paraffin or resin, sectioned, and mounted on glass slides. The paraffine sections were demounted and gradually rehydrated with water. Non-specific binding was blocked using a blocking solution. Specific primary antibodies were added and incubated at room temperature or 4°C overnight. The tissue sections were washed with phosphate-buffered saline (PBS) to remove unbound primary antibodies before incubating with secondary antibodies labeled with enzymes or fluorophores for 12 h. The stain was developed using 3,3′-diaminoben-zidine. Cell nuclei were counterstained with hematoxylin. Staining was observed microscopically.
Immunofluorescence
Paraffin-embedded tissue sections were fixed in 4% paraformaldehyde, dewaxed using xylene, and rehydrated with an ethanol gradient. Antigen retrieval was performed using a citrate buffer. The tissue sections were blocked with serum to reduce non-specific binding. Primary antibodies were added and incubated in a humid chamber overnight at 4°C or for several hours at room temperature. The tissue sections were washed multiple times with PBS to remove unbound primary antibodies before adding diluted fluorescently labeled secondary antibodies and incubating them in the dark. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole, and the tissues were washed to remove excess dye before mounting with an anti-fluorescence quenching mounting medium. Images were captured under a fluorescence microscope.
Statistical analysis
Results are expressed as mean ± standard error of the mean (SEM). An unpaired t test was performed to determine the statistical significance of pairwise differences. Correlation analyses were performed using Pearson's rho (ρ) test. Receiver operating characteristic (ROC) curves were employed to evaluate the predictive power of key genes for AAA. All statistical analyses were performed using SPSS 23.0 software (IBM, Armonk, NY, USA). A p-value <0.05 was considered statistically significant.
Results
DEG identification and analysis
Gene expression data from the aortic samples of 69 patients with AAA and 25 non-AAA controls. Hierarchical clustering analysis indicated that gene expression data clearly distinguished between patients with AAA and healthy individuals (Figure 1a). Among the DEGs identified in the AAA versus healthy individual comparison, 86 were upregulated and 318 were downregulated (Figure 1b). Following analysis in R, the differences between AAA and normal aortic tissue in the GSE7084, GSE47472, and GSE57691 datasets were displayed in a volcano plot (Figure 1c).

Differentially expressed gene (DEG) analysis. (a) Heatmap of 404 DEGs. (b) Downregulated and upregulated genes. (c) Volcano plot of DEGs; red: downregulated genes, green: upregulated genes.
Functional and pathway enrichment analysis of DEGs
The ClusterProfiler package in R was employed for GO enrichment analysis and KEGG pathway analysis to determine the functional roles of the identified DEGs. Among the 404 DEGs, the top biological processes included granulocyte migration, leukocyte migration, and neutrophil migration. The most significant cellular components were tertiary granules, secondary lysosomes, and ficolin-1-rich granule membranes. The molecular functions of genes enriched in the AAA samples were predominantly associated with chemokine activation, chemokine receptor binding, and kinase activation (Figure 2a and b). KEGG pathway enrichment analysis indicated that these DEGs were primarily involved in cytokine–receptor interactions and chemokine signaling pathways (Figure 2c).

GO enrichment and KEGG pathway analysis of upregulated DEGs in patients with AAA compared to non-AAA controls. (a) Bubble plot of GO enrichment analysis. (b) Circular plot of GO enrichment analysis. (c) Bubble plot of KEGG pathway analysis.
PPI network analysis
STRING was used to construct a PPI network for upregulated DEGs, visualized within Cytoscape (Figure 3d). Additionally, the Cytoscape plugin MCODE was employed to identify key modules within the PPI network (Figure 3b). Among the significantly upregulated DEGs, a key module containing 12 nodes was identified (Figure 3e). CXCL8 was the most significantly upregulated gene within the key module (Figure 3c). The top 10 upregulated DEGs in AAA (Figure 3a), the top 10 upregulated DEGs in the interaction degree of the PPI network (Figure 3b), and the DEGs in the key modules (Figure 3c) were applied to construct a Wayne diagram to identify intersecting genes. Four key DEGs encoding CC-chemokine receptor 7, C-X-C motif chemokine ligand 8 (CXCL8), interleukin (IL)-1β, and C-C motif chemokine ligand 3 were identified (Figure 3f).

PPI network analysis. Node color represents the degree of gene interaction. (a) Top 10 upregulated DEGs in AAA. (b) Top 10 upregulated genes with the highest interaction levels in the PPI network. (c) Interaction levels among DEGs in the key module. (d) PPI network for upregulated DEGs identified from datasets GSE7084, GSE47472, and GSE57691. (e) Key modules identified using the MCODE plugin. (f) Intersection of genes from A, B, and C.
Key genes of clinical significance in AAA
The STRING and Gene MANIA database analyses indicated possible interactions between CXCL8 and NLRP3 (Figure 4). In aortic wall tissue from patients with AAA, NLRP3, IL18, and IL1B expression levels were upregulated (Figure 5d). CXCL8 exhibited a consistently significant positive correlation with these three genes (Figure 5a to c). The area under the ROC curve (AUC) suggested that CXCL8 demonstrated potential diagnostic value and could serve as a biomarker for AAA (Figure 5e).

(a) Potential interaction between CXCL8 and NLRP3 suggested by the STRING databases. Network nodes represent proteins. Edges represent protein–protein associations. (b) CXCL8 may interact with NLRP3 based on Gene MANIA browser analysis.

Upregulation of CXCL8 in AAA and its diagnostic value. (a–c) Positive correlation between CXCL8 and the expression of NLRP3, IL-18, and IL-1β, respectively. (d) Upregulation of CXCL8, NLRP3, IL-18, and IL-1β expressions in AAA. (e) ROC curve analysis showing the sensitivity and specificity of CXCL8 for diagnosing AAA.
Role of CXCL8 in regulating NLRP3 inflammasome and aggravating vascular inflammation in AAA
Peripheral blood samples were collected from 12 patients with AAA and 12 non-AAA controls at our center. RT-PCR results showed that the transcription levels of CXCL8 and NLRP3 were upregulated in patients with AAA (Figure 6a and b), exhibiting a significant positive correlation (Figure 6c). The AUC suggested that CXCL8 and NLRP3 demonstrated potential diagnostic value for AAA (Figure 6d and e), with their combined use improving diagnostic accuracy (Figure 6f).

Upregulation of CXCL8 and the inflammatory factor NLRP3 in AAA. (a) Upregulation of CXCL8 expression. (b) Upregulation of NLRP3 expression. (c) Correlation analysis. (d, e) ROC curve analysis of the diagnostic sensitivity and specificity of CXCL8 and NLRP3, with (f) their combined use offering enhanced diagnostic performance.
Immunofluorescence staining of abdominal aortic tissue samples revealed higher CXCL8 and NLRP3 expression levels in the aortic walls of patients with AAA than those in non-AAA controls (Figure 7a and b). Additionally, hematoxylin and eosin (HE) and Van Gieson staining showed that the aortic wall tissue of patients with AAA was disrupted and exhibited reduced elastic fibers with a disorganized tissue structure (Figure 7c and d). These findings align with the changes observed in this study, further indicating that upregulated CXCL8 in AAA aortic tissue may contribute to vascular inflammation, intensify damage to the vascular wall, and accelerate AAA progression.
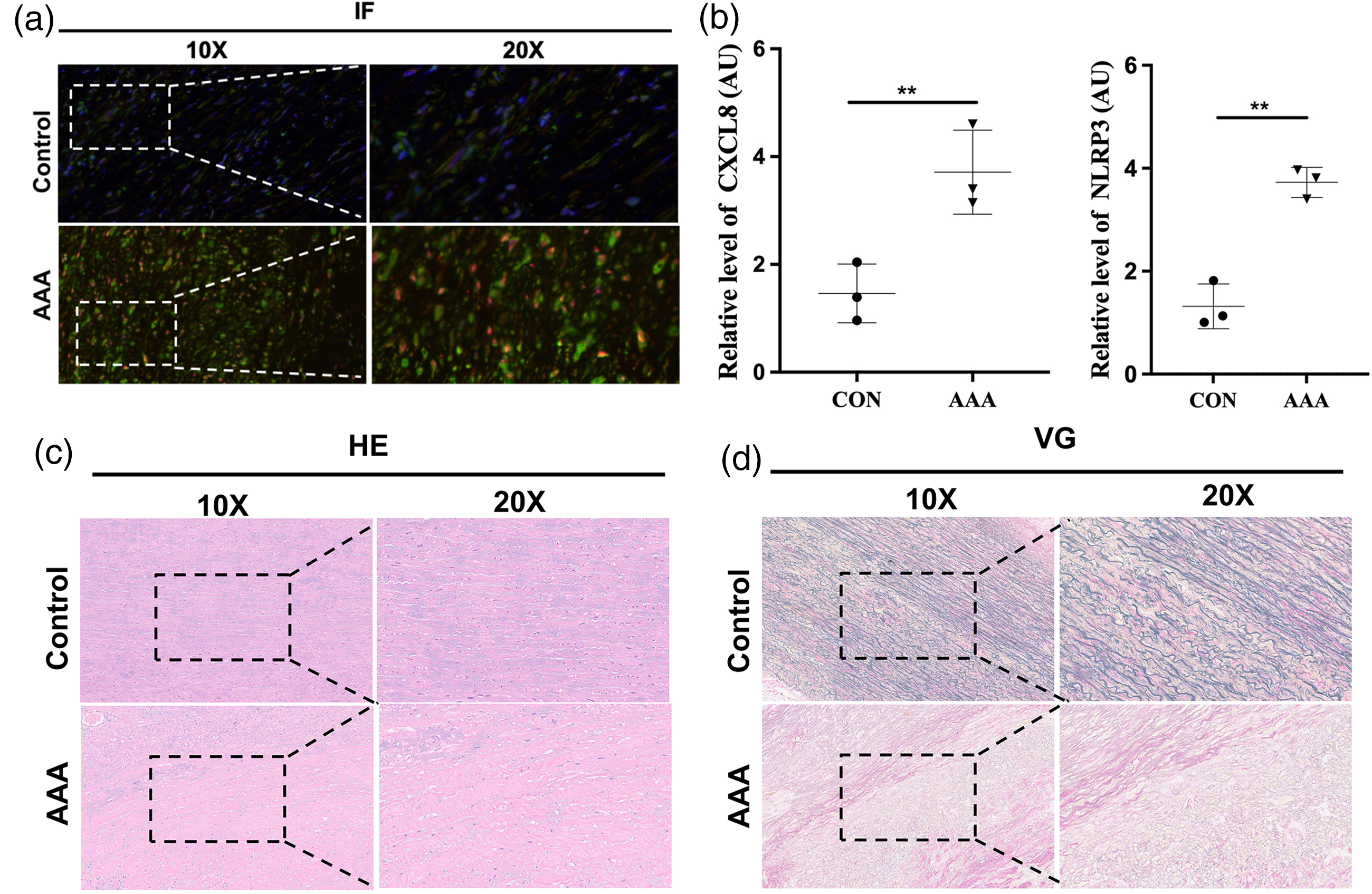
(a, b) Immunofluorescence staining of CXCL8 and NLRP3 in the aortic wall of patients with abdominal aortic aneurysm (AAA) compared to non-AAA controls. (c, d) HE and VG staining results.
Discussion
Inflammatory responses are linked to chemokine function, which mediates the adhesion and aggregation of inflammatory cells in the aortic wall.19,20 In the current study, CXCL8 expression in the aortic walls of patients with AAA was significantly higher than that in those of non-AAA controls. These findings suggest that CXCL8 could serve as a valuable clinical marker for AAA. CXCL8, also known as IL-8, is a multifunctional cytokine primarily produced by monocytes/macrophages and neutrophils. CXCL8 specifically binds its receptors (IL-8RA/CXCR1 and IL-8RB/CXCR2), regulating the expression of various genes with an important role in inflammation. 21 Upon encountering stimuli, such as lipopolysaccharides, tissue-resident monocytes, and macrophages secrete CXCL8, exhibiting a chemotactic effect on circulating neutrophils. Furthermore, CXCL8 can upregulate β2 integrin CD11b/CD18 expression, activating neutrophils and guiding them to the inflammation site. This triggers a respiratory burst in neutrophils, leading to the release of superoxides (O2−, H2O2) and lysosomal enzymes, causing localized inflammation and potentially damaging tissues. 22
The key upregulated genes in patients with AAA were primarily enriched in the chemokine signaling pathway. Iida and Xu 23 demonstrated that in a mouse model of elastase-induced AAA, the peptide inhibitor MKEY effectively inhibited AAA formation by blocking the interaction between CCR5 and its receptor CCR4. MKEY is one of the few drugs with a notable suppressive effect on established AAA. Moran and Jose 24 found that rapamycin/everolimus reduces AngII-induced AAA formation by decreasing the expression of chemokine CCR2. Similarly, Marinkovic and Hibender 25 reported that azathioprine inhibits the expression of various AAA-related chemokines, alleviating endothelial cell damage, suppressing inflammation, and mitigating Ang II-induced AAA formation. Considering these findings, chemokines may mediate inflammatory cell adhesion, aggregation, and infiltration within the aortic wall and regulate inflammatory factors. Thus, inhibiting relevant chemokines in AAA could be a promising approach to slowing or reversing AAA progression.
Inflammation is critical in the development and progression of cardiovascular diseases. We postulate that inflammation induces pyroptosis in VSMCs; the subsequent release of pro-inflammatory cytokines triggers an inflammatory cascade that exacerbates vascular wall damage and accelerates AAA progression. Pyroptosis is a form of programmed cell death that occurs when cells are stimulated by infectious or endogenous damage-related signals and is characterized by an inflammatory response. 26 Pyroptosis exhibits morphological features similar to necrosis and apoptosis. However, unlike apoptosis, pyroptosis is mediated by inflammasomes, causing cell swelling and loss of membrane integrity. This disrupts intracellular and extracellular substance flow and the release of cellular contents, which can trigger and intensify inflammation, potentially inducing an inflammatory cascade. 27 Pyroptosis is also accompanied by the production and release of inflammatory mediators, which can further promote the inflammatory response and pyroptosis. Endothelial, smooth muscle and dendritic cells can undergo pyroptosis. 28 Pyroptosis in VSMCs and the resulting extracellular matrix are closely related to the stability of atherosclerotic plaques and acute cardiovascular events. 29 He and Ma 30 discovered that in hypoxic rat models and pulmonary artery smooth muscle cells grown under hypoxic conditions, glioma-associated homologue-1 expression is upregulated, promoting smooth muscle cell pyroptosis. Haldar et al. 31 revealed that inflammation-induced pyroptosis in bladder smooth muscle cells, accompanied by cell proliferation and hypertrophy, is a primary cause of bladder dysfunction. Moreover, during atherosclerosis (AS), oxidized low-density lipoprotein induces melanoma differentiation-associated protein overexpression in VSMCs, leading to pyroptosis and AS aggravation. 32 Furthermore, various cardiovascular disease risk factors, including lipid metabolism disorders, hypertension, obesity, smoking, diabetes, and metabolic syndrome, can activate the NLRP3 inflammasome, promoting the expression and release of inflammatory mediators, such as IL-1β and IL-18, a process that resembles classic pyroptosis pathways. This suggests that pyroptosis may contribute to the onset and progression of cardiovascular diseases through inflammatory pathways. 33
The inflammasome is a large multiprotein complex that forms in response to pathogen-associated molecular patterns and damage-associated molecular patterns. It drives the maturation of pro-inflammatory cytokines IL-1β and IL-18 and cleaves gasdermin D, inducing pyroptosis. The NLRP3 inflammasome is the most extensively studied and typically comprises NLRP3, the apoptosis-associated speck-like protein containing a C-terminal caspase activation and recruitment domain (ASC), and pro-caspase-1. NLRP3 comprises three domains: a C-terminal leucine-rich repeat domain, a nucleotide-binding and oligomerization domain, and an N-terminal pyrin domain (PYD). ASC contains a PYD and caspase activation and recruitment domain, which interact to form the inflammasome. Once the NLRP3 complex is assembled, it activates caspase-1, triggering IL-18 and IL-1β secretion, initiating inflammation, and triggering pyroptosis. Factors such as neurotoxins, α-synuclein aggregation, mitochondrial reactive oxygen species, and impaired mitophagy can activate the NLRP3 inflammasome in microglia, resulting in IL-1β and IL-18 release, which can mediate pyroptosis in neuronal cells.34,35
The present study analyzed messenger RNA expression data from the aortic samples of 69 patients with AAA and 25 health controls. After merging and normalizing the datasets, bioinformatics analysis was conducted on the upregulated DEGs. CXCL8 was identified as a key DEG predominantly involved in regulating the chemokine signaling pathway. CXCL8 expression in the aortic wall was higher in patients with AAA than in non-AAA controls, with predictive potential in AAA serving as a possible diagnostic gene. Data from the STRING and GeneMania databases indicated a direct or indirect interaction between CXCL8 and NLRP3. In AAA tissue, NLRP3, IL18, and IL1B expression levels were upregulated, and CXCL8 exhibited a consistently significant positive correlation with these three genes. RT-PCR results from peripheral blood samples from 12 patients with AAA and 12 non-AAA controls confirmed that the transcription level of CXCL8 was significantly positively correlated with NLRP3 upregulation in patients with AAA. Immunofluorescence staining revealed increased expression of CXCL8 and NLRP3 in AAA aortic walls. NLRP3 was significant in AAA progression, activating IL-1β and IL-18. 36 Although co-immunoprecipitation analyses were not performed in the current study, STRING and Gene MANIA database analyses indirectly suggested an interaction between CXCL8 and NLRP3 in AAA. Puchenkova et al. 37 identified a unique proteomic signature in patients with AAA, emphasizing changes in proteins associated with inflammation, extracellular matrix remodeling, and VSMC function. These findings are consistent with our transcriptomic data, which highlight the significant role of inflammation in AAA. Their proteomic analysis revealed an increase in matrix metalloproteinases, known to degrade elastin and collagen in the aortic wall, aligning with the upregulation of genes involved in these processes observed in our transcriptomic data. Additionally, staining revealed that elastic fibers in AAA aortic wall tissue were damaged and depleted, with a disorganized tissue structure. These findings suggest that CXCL8 upregulation in AAA aortic tissue may contribute to the inflammatory response and vascular wall damage, accelerating AAA progression.
This study had several limitations. CXCL8 has not been further confirmed by in vitro studies or other functional studies. In addition, the significance of the expression level of CXCL8 and clinical data could not be considered in this study. However, to some extent, our results could be enlightening for subsequent mechanism studies. Our study lacks in-depth mechanistic studies targeting these aspects and to establish a direct link between CXCL8 and NLRP3, more experimental validation is required. This is also the content we plan to further validate through experiments in the future.
Conclusion
We propose that CXCL8 upregulation in the aorta of patients with AAA mediates the infiltration of inflammatory cells in the aortic wall, accelerating AAA progression.
Footnotes
Author contributions
LW and YH contributed to the study conception. XX and GH contributed to data collection. YH, XX, and GH contributed to the analysis. XH, WF, and WL contributed to the investigation. YH contributed to manuscript preparation. YH, XX, and LW contributed to funding acquisition. All authors contributed to the critical review, revision, and final approval of the article. All authors are accountable for all aspects of the work.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (grant number: 81970412), Xiamen Medical and Health Guidance Project (grant number: 3502220214201088), and Xiamen Medical and Health Guidance Project (grant number: 3502220214201062).
Consent to participate
Written informed consent was obtained from all patients.
Consent to publish
Ethics approval
The study was approved by the Institutional Review Board of the Xiamen Branch of Zhongshan Hospital (Ethics Approval Number: B2023-153; Date: 17 January 2024).