
Abstract
Objectives:
Human papillomavirus (HPV) infection is closely related to upper respiratory mucosal lesions, and the most common disease is adult upper respiratory papilloma, which has a certain probability of cancer transformation.This study conducted in vitro tissue and cell experiments to explore the inflammatory mechanisms associated with HPV + adult laryngeal papilloma.
Methods:
We compared differential expression of AIM2 and IL-1β between HPV (High-risk) negative and positive adult laryngeal papilloma patients. In vitro experiments were conducted to investigate the differences in expression of AIM2, Caspase-1, and IL-1β in HPV− and HPV+ upper respiratory mucosal cells.
Results:
The expression level of AIM2 and IL-1β was higher in HPV (High-risk) positive papilloma tissue than HPV (High-risk) negative papilloma tissue. The expression of AIM2, Caspase-1, and IL-1β in HPV+ cells was also significantly higher than in HPV− cells.
Conclusions:
The expression of IL-1β mediated by AIM2 was associated with chronic inflammation of upper respiratory mucosal tissue caused by HPV infection, and it may yet be associated with further pathological changes.
Introduction
Respiratory papillomatosis (RP) is a benign airway tumor caused by human papillomavirus (HPV) infection and is a common HPV-related upper respiratory mucosal disease. In recent years, new endoscopic techniques such as narrowband imaging (NBI) have been developed that have improved the detection rate of adult laryngeal papillomas. The treatment method for this disease is mainly surgery, but RP is prone to recurrence after surgery. RP also often has a severe impact on patient dysphonia and can cause upper respiratory tract stenosis after multiple surgeries. What's worse, 3%–7% of adult laryngeal papilloma cases become malignant. 1 At present, the pathogenesis of this disease has not been fully elucidated, and feasible treatment methods have yet to be proposed. Therefore, an exploration of the pathogenesis of RP with the goal of finding a feasible way to block the progression of papilloma in the early stages is urgently needed. In our previous research, we found that IL-1β exists in HPV-positive laryngeal papilloma tumor tissue, and in this current study we therefore evaluated the expression of IL-1β, AIM2, and Caspase-1 and their specific mechanisms in an attempt to elucidate the regulation of HPV in adult laryngeal papilloma to provide new intervention strategies for the treatment of RP.
Materials and methods
HPV infection and subtype testing
We collected tissue samples from 34 patients with laryngeal papilloma (isolated papilloma) from June 2021 to June 2022 who visited Beijing Tongren Hospital, Capital Medical University. 34 cases of adult laryngeal papilloma tissue were taken for HPV detection using RNA in situ hybridization. The detection probe used was a mixed probe (ZSGB-BIO Crop., Beijing, China), with 18 subtypes of high-risk patient: 16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 73, and 82. The positive criterion was punctate brown signals visible in the nucleus and/or cytoplasm of ≥10% of tumor cells under a light microscope (20×). 2
Immunofluorescence staining
Immunofluorescence positivity was determined as follows. We first observed the slices under a microscope then selected 5 representative fields of view under high magnification and counted 100 cells in each field after collecting the images. The proportion of positive cells in each field of view in the combined slice was then recorded.
Cells and test materials
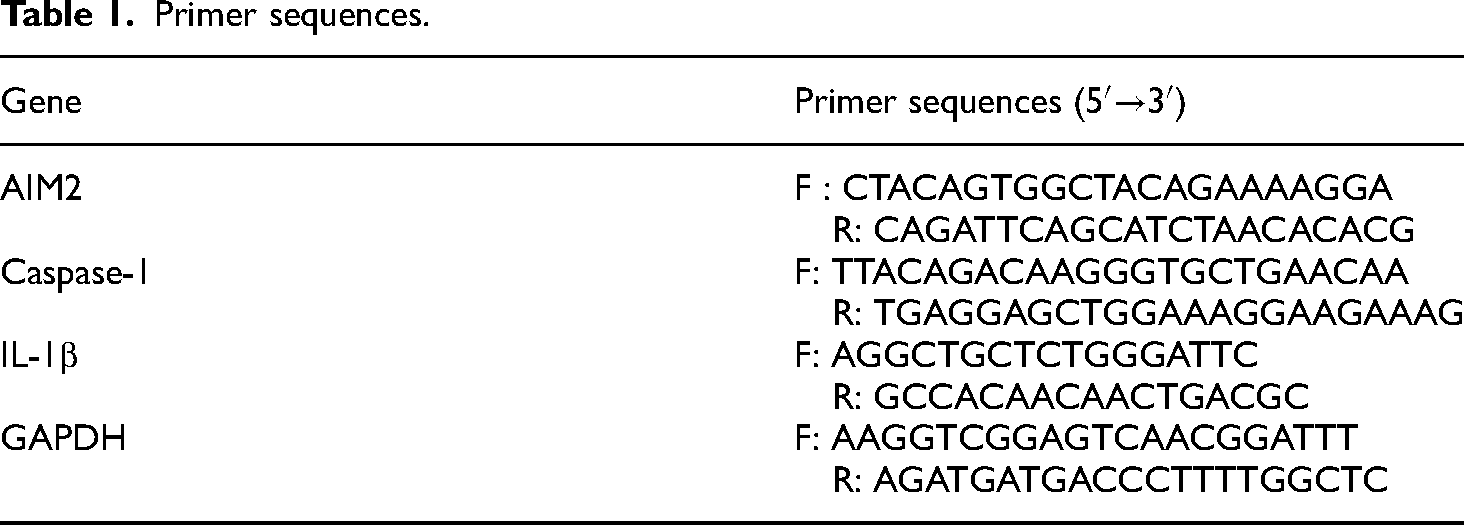
HBE135-E6E7 (ATCC CRL-2741): HPV + human bronchial epithelioid cells (12HBE) were purchased from ATCC, BEAS-2B (ATCC CRL-9609): human bronchial epithelial cells (BEAS) were purchased from the Kunming Cell Bank of the Chinese Academy of Sciences (Kunming Branch of the National Experimental Cell Resource Sharing Platform). A Human IL-1β ELISA Kit by RayBio was used to detect IL-1bβ protein levels. AIM2 shRNA lentivirus was purchased from Shanghai Jikai Gene Medical Technology Co., Ltd., and AIM2 overexpression plasmid was purchased from Beijing Baiaochuan Biotechnology Co., Ltd. Finally, a Lipofectamine 3000 transfection kit was purchased from Thermo Fisher Scientific (China) Co., Ltd., and PCR primers were purchased from Shanghai Biotechnology Services Co., Ltd. (Table 1).
Primer sequences.
RNA extraction and RT-PCR
To perform RNA extraction and RT-PCR analysis, we began by precooling the Trizol reagent on ice, digesting the cells, cleaning the cell precipitate with PBS, and discarding the supernatant after centrifugation. Next, we took 1 mL of pre-cooled Trizol and added it to the cell precipitate. After blowing, we transferred the mixture to an enzyme-free 1.5 mL Ep tube. After this we added 200 µL trichloromethane, thoroughly shake it, mixed the Trizol and trichloromethane, and let it stand on ice for 5 min. This was then centrifuged at 4°C in a low-temperature centrifuge at 12,000 rpm for 5 min. The supernatant was then placed into a new EP tube and treated with an equal volume of isopropanol. This was mixed well and left to stand on ice for 10 min. Then, the mixture was centrifuged again at 4°C and 12,000 rpm for 10 min. The white precipitate was poured out the liquid in the tube, and 1 mL of 70% ethanol was added for thorough washing. One final centrifuge at 4°C and 7500 rpm was then performed for 5 min, and the sample was dried in an ultra-clean workbench.
After this, 20 µL of DEPC water fully dissolved the RNA, and using a 1 µL sample, the RNA concentration was measured using an ultra-micro spectrophotometer. 1 µL out of the extracted RNA was then added to the PCR tube and mixed with 1 µL of Oligo (dT) 18 prime. Deionized water without RNA was used to bring the total volume to 12 µL. Next, the sample was placed in a PCR instrument at 65°C for 5 min and then quickly cooled in an ice box. The following components were then added to the PCR tube in order: 5× Reaction Buffer: 4 µL; RiboLock RNase Inhibitor (20 U/µL): 1 µL; 10 mM dNTP Mi×: 2 µL; and RevertAid M-MuLV RT (200 U/µL): 1 µL to make a total volume of 20 µL. These components were then mixed thoroughly and evenly. Finally, the PCR program was left to react at 42°C for 60 min, 25°C for 5 min, and 70°Cfor 5 min, before being stored at 4°C. The final cDNA product itself was stored at −20°C. The preparation of the reaction system included RT qPCR in the amount of 1 µL, upstream and downstream primer in the amount of 1 µL, SYBR Green in the amount of 4 µL, and ultra-pure water in the amount of 4 µL. After 1 cycle at 95°C, 35 cycles at 94°C, 60°C, 72°C (each), and 1 cycle at 72°C, the resulting sample was stored at 4°C.
ELISA
To carry out our ELISA testing, we first put the kit at room temperature for 60 min for balancing and took out the flats required in the aluminum foil bag. The corresponding positions of standard and sample wells were set, and different concentrations of 50 µL standard samples were added to each well (standard concentration: 125 pg/mL, 250 pg/mL, 500 pg/mL, 1000 pg/mL, and 2000 pg/mL). next, we added an equal volume sample to the sample hole to be tested. 100 µL of HRP-labeled detection antibody was added to the standard well and sample well, and the reaction well was sealed with sealing film and incubated in a 37 37°C incubator for 60 min. After this, the liquid was discarded, and the sample was patted dry on absorbent paper. Detergent was then added and left to stand for 1 min before being shaken off and patted dry again on absorbent paper, a process which was repeated 5 times. We then added 50 µL of substrates A and B to each well and incubated at 37°C in the dark for 15 min before adding 50 µL of termination solution to each well and measuring the OD values at a wave length of 450 nm within 15 min.
siRNA transfection
After 48–72 hour of virus infection (70%–80% fusion degree), the cells were further cultured in a culture medium containing an appropriate concentration of Puromycin. After screening for 48 h, all empty cells and Puromycin groups died, and at this time, all remaining cells in the virus infection group were positive. The concentration of Puromycin was then reduced to a maintenance concentration (1/2–1/4 of the screening concentration), and we continued to screen and amplify the infected cells to collect cells for RT qPCR identification. Finally, we froze and preserved the cells that displayed normal identification results (Table 1).
Plasmid transfection
After amplification and extraction of the constructed plasmid, we used agarose gel electrophoresis and ultraviolet spectrophotometer to detect the concentration and purity before digesting and centrifuging the cells, resuspending them with 1 mL complete medium, counting them, putting 2 × 105 cells into each hole of a six-hole plate, and placing them into a 5% CO2 37°C incubator overnight. We then discarded the cell supernatant, added 1 mL of serum-free medium, and placed the samples it in the incubator for 1 h of starvation. Next, we prepared sterile 1.5 mL EP tubes, labeled them as A and B, and added 250 of µL serum-free medium and 5 µL of Lipo3000 to tube A and 250 µL of serum-free medium, 5 µL P3000, and 5 µG plasmid to tube B. This was mixed well and left to stand for 5 min, after which A and B were mixed and left to stand for 20 min. After removing the 6-well plate, we discarded the supernatant, added 1.5 mL of complete culture medium, and then evenly added the mixed solution to the well. This was then incubated for 24 h before carrying out RT qPCR validation of transfection efficiency.
Statistical methods
All laboratory testing content was repeated in triplicate. The data were then expressed as mean ± standard deviation prior to analysis. If the variance between the means was uniform, a paired t-test is used. If the variance was not uniform, rank sum test was used. Differences were assumed to be statistically significant if p < 0.05. SPSS 24.00 software was used for all statistical analysis, and GraphPad Prism 9.0 software was used to draw statistical charts.
Ethical approval
This study was approved by the Ethics Committee of Beijing Tongren Hospital (04/07/2022; TRECKY2021-050). All patients involved signed informed consent forms, and the study was conducted in accordance with the Helsinki Declaration of 1975 as revised in 2013.
Results
Clinical data
Among the 34 patients included in the study, there were 27 males and 7 females, and there were 14 HPV-positive (High-risk) patients and 20 HPV-negative (Non-high-risk) patients. We found a significant difference in the positivity rate in different groups, and the positive rate was significantly higher for AIM2 and IL-1β in the HPV-positive (High-risk) group (p = 0.000, p = 0.042) (Table 2, Figure 1A and B).

(A, B) The immunofluorescence positive expression of IL-1β and AIM2 in HPV (High-risk) positive and negative papilloma tissues. *p = 0.042, ****p = 0.0000. (C) Immunofluorescence microscopy appearance (40×): IL-1β in HPV-positive (High-risk) and HPV-negative (Non-high-risk) tissues.
Comparison of intergroup differences between HPV (high-risk) positive group and negative group patients.

RT-PCR and ELISA detection of AIM-2, Caspase-1, and IL-1β expression in HPV+ and HPV− cells
The results from using real-time quantitative PCR to detect AIM-2, Caspase-1, and IL-1β mRNA expression in HPV-positive and HPV-negative cells are shown in Figure 2A. There was a significant difference in mRNA levels between HPV-positive (16HBE) and HPV-negative cells (BEAS), and this difference was statistically significant (p = 0.001, p = 0.0037, p = 0.0128). Further collection of two cell supernatants for ELISA detection of IL-1 β protein levels, showed that IL-1β levels between HPV-positive (16HBE) and HPV-negative cells (BEAS) were also significantly different (p < 0.0001; Figure 2B). This indicates that HPV infection significantly affects the secretion level of IL-1β and interacts with the expression of AIM-2 and Caspase-1.

(A) Real-time fluorescence quantitative PCR detection of the expression of AIM-2, Caspase-1, and IL-1β mRNA in HPV-positive and HPV-negative cells. ***p = 0.001, **p = 0.0037, *p = 0.0128. (B) ELISA detection of IL-1β protein levels in HPV-positive and HPV-negative cells. ****p < 0.0001. (C) Real-time fluorescence quantitative PCR detection of the expression of AIM-2, Caspase-1, and IL-1β mRNA in HPV-positive and HPV-negative cells transfected with AIM-2 siRNA. ****p < 0.0001, **p = 0.0120, **p = 0.0062; ns p = 0.1559, ns p = 0.0584, ns p = 0.1357. (D) ELISA detection of the protein levels of IL-1β in HPV-positive and HPV-negative cells transfected with AIM-2 siRNA. ****p < 0.0001, ns p = 0.0541. (E) Real-time fluorescence quantitative PCR detection of the expression of AIM-2, Caspase-1, and IL-1β mRNA in HPV-positive and HPV-negative cells transfected with pEGFP-hAIM-2. *p = 0.0163, **p = 0.0022, **p = 0.0074; ns p = 0.3408, ns p = 0.4182, ns p = 0.1248. (F) ELISA detection of the protein levels of IL-1β in HPV-positive and HPV-negative cells transfected with pEGFP-hAIM-2. ***p < 0.0001, **p = 0.0078.
AIM-2, Caspase-1, and IL-1β expression after transfection with AIM-2 siRNA to knockdown AIM2
Real-time quantitative PCR was used to detect AIM-2, Caspase-1, and IL-1β in HPV-positive and HPV-negative cells transfected with AIM-2 siRNA 48 h after transfection, as well as in untransfected control cells. The mRNA expression results are shown in Figure 2C. After transfection with AIM-2 siRNA, the AIM-2 mRNA levels in HPV-positive and HPV-negative cells significantly decreased (p < 0.0001, p = 0.0120). In addition, there was a significant decrease in Caspase-1 mRNA levels between the two groups of cells, but the difference was not statistically significant in the HPV-positive cells (p = 0.1559, p = 0.0062). Although there was a difference in IL-1β mRNA expression between HPV-positive (16HBE) and HPV-negative cells (BEAS), this difference was not statistically significant either (p = 0.0584, p = 0.1357).
Further collection of supernatants from two groups of cells with AIM-2 siRNA and control cells was carried out in order to detect the protein levels of IL-1β. As shown in Figure 2D, there was a significant difference in the levels of IL-1β between HPV-positive (16HBE) and HPV-negative cells (BEAS). The protein levels of IL-1β were significantly decreased in cells infected with AIM-2 siRNA, and the difference was statistically significant in HPV-positive cells (p < 0.0001) but not in HPV-negative cells (p = 0.0541) (Figure 2D). This is likely related to the low basal level in HPV-negative cells. Overall, however, these results indicate that AIM-2 affects the expression of Caspase-1 and IL-1β.
AIM-2, Caspase-1, and IL-1β expression after pEGFP-hAIM-2 transfection
Real-time quantitative PCR was also used to detect the mRNA expression of AIM-2, Caspase-1, and IL-1β in HPV-positive and HPV-negative cells transfected with pEGFP-hAIM-2 48 h after transfection, as well as in untransfected control cells. These results are shown in Figure 2E. After transfection with pEGFP hAIM-2, the AIM-2 mRNA levels in HPV-positive and HPV-negative cells significantly increased (p = 0.0163, p = 0.0022). In addition, there was a significant increase in Caspase-1 mRNA levels between the two groups of cells, but the difference was not statistically significant in HPV-positive cells (p = 0.3408, p = 0.0074). Although there was a difference in IL-1β mRNA expression between HPV-positive (16HBE) and HPV-negative cells (BEAS), this difference was also not statistically significant (p = 0.4182, p = 0.1248).
Further collection of supernatants from two groups of cells transfected with pEGFP-hAIM-2 and control cells to detect the protein level of IL-1β showed that there was a significant difference in the level of IL-1β between HPV-positive (16HBE) and HPV-negative cells (BEAS) transfected with pEGFP-hAIM-2, and the difference was statistically significant (p < 0.0001, p = 0.0078) (Figure 2F). Thus, AIM-2 affected the expression of Caspase-1 and IL-1β after overexpression of AIM-2.
Discussion
IL-1β is an important member of the IL-1 family as it induces the release of inflammatory mediators and related chemokines by immune cells, which can lead to inflammatory diseases. 3 However, IL-1β also plays a beneficial role in acute inflammation and initiating adaptive anti-tumor immune response. In the process of chronic inflammation, though, IL-1β has a promoting effect on tumor development. 4 Inflammasomes are multiprotein complexes assembled by intracellular pattern recognition receptors (PRRs) and are important components of the natural immune system. Inflammasomes can recognize pathogen-related molecular patterns to recruit and activate the pro-inflammatory protease Caspase-1. Activated Caspase-1 cleaves IL-1β precursor, and IL-18 produces mature cytokines. At present, there are four main types of inflammasomes, namely NLRP1, NLRP3, IPAF, and AIM2. Among them, AIM2 can promote pro-IL-1 in the innate immune process by regulating the activation of Caspase-1, which cleaves pro-IL-1β into mature IL-1β.5,6 In our previous research, we found high expression of IL-1β, AIM2, and Caspase-1 in HPV-positive laryngeal papilloma tumor tissue. Additionally, a positive correlation between AIM2 expression and malignant transformation and recurrence of papilloma has already been reported. 7 So, this study further explores the correlation between HPV, AIM2, and IL-1β in vitro experiments.
AIM2 is currently the only known inflammasome that can recognize cytoplasmic DNA, and it generally senses the dsDNA of pathogens within the cytoplasm. 8 The HPV pathogen takes the form of dsDNA, and this may promote increased expression of IL-1β by activating AIM2 inflammasomes. The biological activity of IL-1β is mainly regulated at the transcriptional and post-transcriptional levels, where the former relies on TNF α to activate NF-κB and then promotes IL-1β transcription. The latter cleaves pro-IL-1 via Caspase-1 proteolytic enzyme into active IL-1β. 9 More specifically, the HIN-200 domain of the AIM2 inflammasome can sense and bind to dsDNA released in the cytoplasm by HPV. 10 A study of macrophages in AIM2-deficient mice found that AIM2 inflammasomes can sense the stimulation of cytoplasmic DNA and specifically sense the DNA released into the cytoplasm by cowpox virus and intracellular bacteria. AIM2 inflammasomes can also monitor DNA damage and respond early, secreting IL-1β and IL-18, while inducing Caspase-1-dependent cell pyroptosis to destroy infected macrophages, thereby limiting the spread of infection. Our current study additionally showed that HPV induces IL-1β expression in upper respiratory mucosal cells by regulating the AIM2/Casase-1 pathway.
In recent years, there has been progress in research on HPV-related tumors and IL-1β. Many studies have shown that in chronic inflammation caused by HPV infection, sustained IL-1β expression may promote the occurrence and progression of tumors. 11 For example, increased expression levels of IL-1β have been observed in tandem with the progression of cervical intraepithelial neoplasia, indicating that the local Th2 inflammatory response in the cervical region is enhanced in this case, which is conducive to the immune escape of HPV virus infection.12,13 Some studies have also shown that HPV can cause AIM2 to release IL-18 and IL-1β in human keratinocytes, 14 and others have found that compared to normal healthy skin, the expression of AIM2, IL-1β, and Casepase-1 was significantly increased in HPV-positive skin lesions. 14 For example, research shows that in human keratinocytes, IL-1β levels significantly increase after immune stimulation or malignant transformation. 15
It can also activate the promoters of oncogenes and immunosuppressive genes in HPV-infected keratinocytes, promoting tumor development. 16 Furthermore, silencing IL-1β with lentivirus has been shown to inhibit the growth of oral squamous cell carcinoma significantly in both in vivo and in vitro experiments, indicating that IL-1β has a certain promoting effect on squamous cell carcinoma caused by HPV. 17 This study preliminarily explored the mechanism of AIM2/IL-1β in causing the disease progression of HPV by measuring the correlation between HPV high-risk types and AIM2/IL-1β and investigating the role of AIM2/IL-1β in both HPV-positive and negative cells. In doing so, we provided an explanation for the immune inflammatory mechanism that underlies the progression of adult laryngeal papillomatosis, providing new directions and laboratory support for clinical treatment.
Limitations
This study has several limitations. First, it is based on a small sample size and needs to be validated by a larger sample. The use of HPV testing kits may result in detection bias and require further validation with larger data. Second, the results of in vitro experiments also need further validation in vivo animal experiments. In the future, we intend to expand our research in both of these ways.
Conclusion
This study shows that IL-1β is mediated by AIM2 in HPV + adult laryngeal papilloma. Moreover, the elevated involvement of IL-1β in the chronic inflammatory state caused by HPV may be associated with further pathological changes. Our results also elucidate the relationship between HPV infection and AIM2/IL-1β. We hope this work will provide laboratory support for further exploration of HPV + adult laryngeal papilloma in the future.
Footnotes
Acknowledgements
Author contributions
G.Y. and W.G. wrote the manuscript. X.C., Z.Y., and Z.H. designed the article. All authors have reviewed the manuscript and approved its publication.
Data availability
All data used in this study are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (Grant Nos. 82071032 and 82072997). Beijing Hospitals Authority Clinical medicine Development of special funding support, code: ZLRK202304.