
Abstract
The amphipathic nature of helical proteins is crucial to their binding features across a broad spectrum of physiological examples, including heat-shock proteins and hyaluronic acid (HA) receptor binding. By taking advantage of the amphipathic balance of amino acids and their presentation in helical faces, novel synthetic peptides can be designed to improve biofunctionality. We present a new approach for designing synthetic alpha helical peptides using a multifaceted analysis, which allows for new bioengineering designs of amphipathic alpha helices. Amphipathic helical peptides were presented with distinct hydrophobic and hydrophilic faces; two series of analogs, namely, peptides AX9 and AX7, were designed to contain a hydrophobic and hydrophilic face, respectively. The presence of one series of peptides exhibited a distinct hydrophobic face and the second series exhibited a distinct hydrophilic face, which was corroborated with reversed-phase chromatography (C8). Using a multifaceted approach to analyze the potential faces of an amphipathic helix, we demonstrated that these helices contain seven distinct “side-viewed” helical faces (based on the hydrophobic face of the AXP series of analogs), which provides additional spatial dimensional information beyond the averaging effect of the hydrophobic moment generated from the “top-down” view of a helical wheel. Furthermore, we cross-compared our recently published HA-binding peptide in this manner to demonstrate that the most significant binding was related to (1) balanced amphipathicity and (2) a distribution of the key HA-binding domain B1(X7)B2 presented spatially. For example, our most effective peptide binder 17x-3 has five of seven faces with B1(X7)B2 domains, while the positive control mPEP35 has three, which reflects a lower affinity. With such a tool, one is able to map helical peptides on an additional dimension to characterize and redesign fundamental amphipathic properties among other critical characteristics, such as sugar and glycan binding, which is a fundamental characteristic feature of cellular interactions in almost every biological system.
Introduction
Amphipathic helices (AHs, or amphipathic helix—AH) are a common secondary structural feature in many natural proteins. Some examples include an 18-residue segment from a protein found in the bacterium Penicillium chrysogenum (PEX11), residues 1–38 in the human protein (GMAP210), heat-shock protein (HSP)-12 containing four AHs, human perilipin (PLIN4) containing a 33 residue repeat peptide, 1 antimicrobial peptides (AMPs), e.g. LL-37 (also known as hCAP18), an amphipathic α-helical peptide, 2 phenol soluble modulin alpha-3 (PSMα3) 3 and receptor for hyaluronic acid-mediated motility (RHAMM)-binding domain I. 4 The characteristic feature of an AH is the presence of a hydrophobic and hydrophilic face, made up of mostly hydrophobic residues (e.g. Ala, Val, Ile) and hydrophilic residues (e.g. Ser, Thr, Lys), respectively. The hydrophobic face of an α-helix within a protein will be oriented toward the hydrophobic core of the protein consisting of a hydrophobic molecular interface(s), whereas the hydrophilic face will be orientated away from the hydrophobic core and be solvent-exposed. The AH is a core structure of many sugar-based interactions including hyaluronic acid (HA), DNA/RNA, and peptidoglycans in bacterial walls. As such, understanding the nature of AH could allow for the emergence of many biotechnological and clinically relevant therapeutic tools.
Synthetic α-helical peptides can be designed to be either amphipathic (AX), i.e. containing both hydrophobic and hydrophilic parts, or nonamphipathic (naA), i.e. does not contain distinct hydrophobic and hydrophilic parts, depending on the intended purpose of the helical peptide.
5
In the case of peptides AX9 and naA, each peptide consists of the same amino acid composition, yet they exhibit different physicochemical characteristics. Since the naA peptide does not have a distinct hydrophobic or hydrophilic face, its interaction with a nonpolar macroscopic interface (such as a reversed-phase chromatography column) will be different from that of the amphipathic peptide AX9. It should be noted that a hydrophobic face does not necessarily consist of only hydrophobic residues but rather is composed of amino acid residues that are predominantly hydrophobic. The converse is true; hydrophilic faces are not necessarily composed of only hydrophilic residues but rather predominantly hydrophilic residues. This relationship of a peptide consisting of distinct faces can be measured by a physicochemical property termed the hydrophobic moment, (

RHAMM-binding domains I and II contain HA-binding domains defined by the formula B(X7)B, where “B” represents a positively charged residue and “X” represents any of the common amino acids, with the exception of negatively charged residues. 11 It should be noted that B(X7)B can also be designated at B1(X7)B2 in order to distinguish between the first and second positive residue of any particular HA-binding domain. The B1(X7)B2 designation will be used throughout this article. When the HA-binding peptide mPEP35 4 was plotted on a helical net, i.e. a two-dimensional x-y plot depicting the cylindrical surface of the α-helix, it was determined that the B1(X7)B2 domains were always oriented on one particular diagonal of the helical net, defined by the formula B1 + 8 = B2. This is a defining feature used to engineer new HA-binding peptides. Additionally, it has been suggested that HA binds to RHAMM-binding domains through B1(X7)B2 domains that are presented on the helical face of an α-helix. 12 Ziebell and Prestwich 13 suggest that residues K553 and K560, which are in RHAMM-binding domains I and II (corresponding to positions K16 and K23 in mPEP35, respectively), interact through salt bridges with the carboxyl groups of glucuronic acid (GlcUA) 14 in HA. These salt bridges contribute ∼40kJ/mole per bridge, which stabilizes the peptide/HA complex. Furthermore, residue V559 (a Val residue in RHAMM-binding domain II) interacts through a hydrophobic interaction (based on the proximity of the two groups, which suggests exclusion of H2O, suggesting a hydrophobic interaction) with a methylene group, which contains two hydrogen atoms covalently bonded to a carbon atom [i.e. – CH2 –] on N-acetylglucosamine (GlcNAc) 14 and contributes on average ∼15 kJ/mole to the stability of the RHAMM/HA complex). 13
Naturally occurring peptides can be re-designed for the purposes of being more effective in a particular applications, e.g. a 20-residue peptide from human apolipoprotein E, designated as A2 (LRKLRKRLLRLRKLRKRLLR), was redesigned by replacing the Arg residue with Trp at reside #12 (LRKLRKRLLRLWKLRKR) which demonstrated efficient cell membrane penetration upon forming an amphipathic helix. 15 We employed a similar philosophy to redesign HA-binding peptides based on B1(X7)B2 HA-binding domains that would demonstrate enhanced HA binding and specificity.
Recently, a series of HA-binding peptides were engineered to be robust HA binders and to exhibit varying degrees of specificity, 16 relative to the positive control peptide mPEP35. 4 Four of the target peptides demonstrated a range of <μH > values, i.e. 0.234 (BHP3), 0.210 (BHP4), 0.199 (17x−3) and 0.072 (Peptide #4) (Figure 1); thus, suggesting a range of amphipathicity or stated another way as a degree of amphipathicity. The four target peptides are helical by circular dichroism (CD) in the presence of trifluoroethanol (20% TFE). To put these <μH > values into perspective, an amphipathic cell-penetrating peptide based on the N-terminal glycosaminoglycan-binding region of human apolipoprotein E 15 has a <μH > of 0.570, RHAMM-binding domain I is 0.453 and peptide AX9 is 0.371 (Figure 1). In the same manner that AHs demonstrate a hydrophobic and hydrophilic face, so do peptides 17x-3, #4, BHP3, and BHP4. Although, based on their <μH>, they demonstrate a much lower degree of amphipathicity than the peptides in the range of 0.570 to 0.371.
Two engineering perspectives were used to approach the design of new HA binders, specifically (i) valency, i.e. increased number of positive charges by introducing additional B1(X7)B2 domains for increased interaction with HA, and (ii) analyzing the multifaceted characteristic of a peptide with a low degree of amphipathicity (alternatively stated, a low
Materials and methods
High-performance liquid chromatography (HPLC) water and acetonitrile were purchased from J.T. Baker (Phillipsburg, NJ, USA). Orthophosphoric acid and triethylamine were purchased from Anachemia (Toronto, Ontario, Canada), while trifluoroacetic acid (TFA) was acquired from Sigma Aldrich (Milwaukee, WI, USA).
Peptides were synthesized using an Applied Biosystems Peptide Synthesizer and eluted as noted in previous work.5,17 Boc chemistry was chosen and a co-poly (styrene-1% divinyl benzene) benzhydrylamine-hydrochloride resin was used. Single coupling was performed as symmetrical anhydrides on all residues except Arg, Asn, and Gln with the use of a hydroxybenzotriazole active ester. All peptides were acetylated with 25% acetic anhydride/dichloromethane (v/v) (10 min), and cleavage/deprotection was performed with anhydrous hydrogen fluoride (20 ml/g resin) with 10% anisole and 2% 1,2 ethanedithiol (1 h at −4°C). Purities were assessed with HPLC (Varian Vist Series 500 chromatograph/HP1090 liquid chromatograph) with a Pep-S C18/C8 column 250 × 4 mm I.D., 5 μm particle size, 100 Å and mass spectrometry (Biolon 20 plasma desorption time of flight mass spectrometer). Peptide elutions were performed as follows: Eluent A is 0.1% aqueous TFA, and eluent B is 0.1% TFA in acetonitrile; peptide separation effected using a linear AB gradient of 1% acetonitrile/minute and a flow rate of 1 mL/min. Column: C8 stationary phase, 300 Å pore size, 7 μm particle size, 220 × 4.6 mm.
Hydrophobicity is designated as < H>, and hydrophobic moment is designated <μH>, consistent with HeliQuest designations (9). Molecular models of helical peptides were performed using structural predictions and presented in GROMACS software.
Experimental design of peptides with a hydrophobic or hydrophilic face
With respect to the first objective, i.e. to increase valency, an additional B1(X7)B2 domain was introduced relative to the control peptide mPEP35, which has been discussed previously. 16 Concerning the second objective, previous work done on defining a hydrophobic and hydrophilic face of amphipathic α-helical peptides using two series of 18 residue peptides designated AX9 and AX7, which are analogs 5 was utilized to develop the criteria for demonstrating the characteristics of a peptide containing a hydrophobic and hydrophilic helical face.
Peptide AX9 (Figure 2) was used to define what constitutes a hydrophobic helical face. The hydrophobic face consists of six Ala residues centered by a “guest residue” in the middle of the hydrophobic face. The primary amino acid sequence for a hydrophobic face is based on a 3-1-3; 3-1-3 repeat. For example, starting with residue 2: 2 + (3) = 5, 5 + (1) = 6, 6 + (3) = 9; 9 + (3) = 12, 12 + (1) = 13, 13 + (3) = 16; thus, hydrophobic residues are placed at position numbers 2, 5, 6, 9 (guest residue), 12, 13 and 16 in the primary sequence. This mathematical formula will allow one to determine the placement of hydrophobic residues in the primary sequence, which will present as a hydrophobic face. Each position in the helical net will define a specific helical diagonal. We have termed these diagonals a seven-residue diagonal, based on the following formula: residue #1 + 7 = residue #2, in order to distinguish it from the HA-binding diagonals defined by B1 + 8 = B2. Furthermore, this arrangement defines a face that will present residues B1 and B2 (of B1(X7)B2) on the same “side” of the α-helix and those two residues will be on a specific seven-residue diagonal. Presentation of these B1 and B2 residues on the same face of an α-helix is identical to that suggested by Day (1998), in his Figure 2b, where B1 and B2 are equal to Day's B1 and B9. Note that in the AX9 model, A5 = B1 and A13 = B2 and these two residues are contained in the “boxed region” of the helical wheel and furthermore are separated by three “neighboring” residues (A2, X9, A16), as depicted by Day's model. 18 This is depicted on the helical net by drawing two solid lines enclosing the residues within a helical face. Note: the “7” in the above formula represents the displacement between the first and second residue in the linear sequence, not the number of amino acids in the helical face. A more specific description of helical net diagonals is: Residues A6 and A13 define a diagonal (Note: 13–6 = 7), A2, X9, A16 define a second diagonal, and A5 and A12 define a third diagonal. Thus, any diagonal consisting of residues displaced by seven residues will define the position in the primary sequence of the next residue in that diagonal. Any such three adjacent diagonals will define a hydrophobic face on an α-helix. In reference to peptide AX9, all the residues to the left of the left-hand line and to the right of the right-hand line will then constitute the hydrophilic face of the peptide and are composed of all hydrophilic residues, i.e. Glu and Lys.

AX9 series of amphipathic α-helices exhibiting a hydrophobic face encased by the two gray lines (including a gray shading) on the helical net. The hydrophobic face is composed of three adjacent seven-residue diagonals, i.e. (A6, A13), (A2, X9-guest residue, A16) and (A5, A12), where a seven-residue diagonal is defined by the formula residue #1 + 7 = residue #2 in the linear sequence. Position AX9 is in the center of the hydrophobic face. The residue markers colored gray, Ala, are in the hydrophobic face and the blue (Lys or Arg) and red (Glu) are in the hydrophilic face of the peptide. The AX7 series of amphipathic α-helices exhibiting a hydrophilic face encased by the two gray lines (including gray shading) on the helical net. The hydrophilic face is composed of four adjacent seven-residue diagonals, i.e. (E1, E8, E15), (K4, K11, K18), (X7-guest residue, K14), and (E3, E10, E17). Position AX7 is surrounded by hydrophilic residues, Glu and Lys. Representative helical wheels and models are shown with highlighted faces.
Regarding designing a hydrophilic face, peptide AX7 (Figure 2) defines what constitutes a hydrophilic helical face. The primary amino acid sequence for a hydrophilic face is based on a 2-1-3-1; 2-1-3-1 repeat. For example, starting with residue 1: 1 + (2) = 3, 3 + (1) = 4, 4 + (3) = 7, 7 + (1) = 8; 8 + (2) = 10, 10 + (1) = 11, 11 + (3) = 14, 14 + (1) = 15; 15 + (2) = 17 and 17 + (1) = 18; thus, hydrophilic residues, Lys and Glu, are placed at position numbers 1, 3, 4, 7 (guest residue), 8, 10, 11, 14, 15, 17, and 18 in the primary sequence. This arrangement also defines a face which will present those eleven residues on the hydrophilic face of the α-helix. This is depicted on the helical net by drawing two solid lines enclosing those eleven residues. It is further noted that residues E1, E8, E15 define a diagonal, K4, K11, K18 define a second diagonal, for a total of four diagonals. Thus, any diagonal consisting of residues displaced by seven residues will define the position in the primary sequence of the next residue in that diagonal, and any such four adjacent diagonals will define a hydrophilic face on an α-helix. In reference to peptide AX7, all the residues to the left of the left-hand line and to the right of the right-hand line will then constitute the hydrophobic face and are composed of all hydrophobic residues, i.e. Ala.
Regarding the design of the AX9 and AX7 peptides, each series of peptides has a “guest residue” at position X9 in the AX9 series and at position X7 in the AX7 series of peptide analogs. The “X” represents each one of the 20 common amino acids, for a total of 20 peptides each for the two sets of peptides analogs.
Experimental evidence demonstrating the presence of a hydrophobic or hydrophilic face
The amphipathic α-helical peptides AX9 and AX7 were subjected to reversed-phase chromatography (RP-HPLC) to demonstrate the existence of a distinct hydrophobic or hydrophilic face. Both series of peptides were chromatographed on a column made of a C8 stationary phase, presenting a very hydrophobic macroscopic interface. Figure 3A demonstrates a large range of retention times (12.1 min; 29.4{I} – 17.3{N}, data reported in Figure 3B) and a relatively good separation of the peptides within this series of analogs. 5 This is a result of the hydrophobic face of AX9 primarily interacting with the stationary phase, and the differences between the elution positions of each peptide in the series are due to the “guest residue” in the center of the hydrophobic face of these peptides. Since there is a relatively large difference in the hydrophobicity for each of the side chains of the amino acids in the guest residue, this results in a significant difference in retention times for this series of amphipathic α-helical peptide analogs.

HPLC profile of the (A) AX9 and (C) AX7 series of amphipathic α-helical peptides. The AX9 series of peptides contain a hydrophobic helical face which contains six Ala residues, with a “guest residue” at position 9 of the primary sequence of the peptide and position 9 is in the middle of the hydrophobic face. The first eluted peptide at 14.4 min is non-amphipathic peptide (naA). The AX7 series of peptides contain a hydrophilic helical face which contains 10 hydrophilic residues (Glu and Lys), with a “guest residue” at position 7 of the primary sequence of the peptide and position 7 is directly surrounded by four hydrophilic residues and an additional six hydrophilic residues in the hydrophilic face. Due to the poor separation of the peptide analogs, the following amino acids are not presented: Cys, Ser, Lys, and Glu. The elution conditions are: Eluent A is 0.1% aqueous TFA and eluent B is 0.1% TFA in acetonitrile; peptide separation effected using a linear AB gradient of 1% acetonitrile/minute and a flow rate of 1 mL/min. Column: C8 stationary phase, 300 Å pore size, 7 μm particle size, 220 × 4.6 mm. The (B) table contains tR retention time in minutes, and ΔRT(G), which refers to the retention time of an analog minus the retention time of the Gly analog. Hydrophobicity is designated as < H > and hydrophobic moment is designated <μH>, consistent with HeliQuest designations. 9
Figure 3C demonstrates a very different elution pattern, i.e. the range of retention times is relatively small (2.3 min; 26.3{L} – 24.0{G}, data reported in Figure 3B) 5 compared to the range of retention times for the AX9 series of peptide analogs. This is due to the fact that the hydrophobic face of AX7 (containing all Ala residues) will be interacting primarily with the hydrophobic stationary phase, and this is evidenced by the relatively similar elution times for both series of peptides, i.e. approximately ∼17–29 min for the AX9 series and ∼24–26 min for the AX7 series. Since there is some chromatographic separation between the peptides of the AX7 series, this means that the hydrophilic face of the AX7 peptides is interacting with the stationary phase to a certain extent based on the differences in hydrophobicity of each of the residues in the guest position of the hydrophilic face of the α-helical peptide. Both sets of chromatographic profiles are evidence of distinct hydrophobic and hydrophilic faces in these amphipathic α-helical peptides. Especially when compared to the low <μH> (0.010) naA peptide, which had a retention of 14.4 min, which was 2.92 min earlier than the first eluted AX9 peptide (AN9). It should be noted that multifaceted analysis of naA indicates that each of the seven helical faces of naA (i.e. naA-i to naA-vii) contain 3 Ala residues (data not shown). Both series of peptides are amphipathic α-helical peptides, which contain both a hydrophobic and a hydrophilic face, and this same principle of a face can be applied to peptides that have a low <μH > . Thus, an α-helical peptide does not need to have a high degree of amphipathicity in order to present a face, the only requirement being that they are α-helical.
Lastly, Figure 3 reports three additional variables in the associated table, i.e. ΔRT(G), which refers to the retention time of an analog relative to the retention time of the Gly analog, <H>, which represents a hydrophobicity for each of the common amino acids when the guest residue is in the center of the hydrophobic face and <µH>, the hydrophobic moment. Furthermore, all the amino acids < H > values represent a hydrophobicity scale. This scale is internally consistent with the scale of Sereda et al., 17 representing a validation of the reverse-phase results obtained in the chromatographic profiles generated in these experiments. Similarly, a scale was generated for the AX7 series of peptide analogs. It is noted that the scale is somewhat different, i.e. the hydrophobicity of an amino acid side chain is dependent on its surrounding environment. More specifically, an amino acid side chain can have a range of hydrophilicity or hydrophobicity, i.e. a maximum and minimum relative hydrophobicity and, similarly, a maximum and minimum relative hydrophilicity. Lastly, the <μH > for each peptide analog is reported. It can be seen that the AX9 series of analogs has a different range of <μH > values (i.e. 0.479–0.298, and a median of 0.369) than that of the AX7 analogs (i.e. 0.372–0.211, and a median of 0.307), which is an expected outcome based on the fact that the hydrophobic face of the AX7 peptide analogs will be interacting with the hydrophobic stationary phase the majority of the time. Additionally, it should be noted that there is an overlap of the two different ranges of <μH>, and this is expected because the predominant hydrophobic face in both series of analogs consists of all six Ala (AX9) residues compared to seven Ala residues (AX7).
Extension to the analysis of HA-binding peptide mPEP35, the positive control (Figure 4)
The mathematical formula for defining the hydrophobic face of the AX9 series of peptides can be used to define such a face in any α-helical peptide. In terms of peptides with a low <μH>, which display a low degree of amphipathicity, there is no distinct hydrophobic (or hydrophilic) face but rather a diffuse array of amino acids of varying degrees of hydrophobicity oriented around the “cylindrical” surface of the α-helix. Since any three adjacent diagonals on the helical net are a “potential helical face” presented by the α-helix to a molecular or macroscopic interface, as described by Dalgicdir et al. 19 and Lu et al., 20 there is more than one face for an α-helix, and they are the same as that described for the hydrophobic face of peptide AX9 series (Figure 2, AX9). In fact, there are seven different faces (each composed of three 7-residue diagonals) presented by the α-helix. Upon rotation of the α-helix by one diagonal at a time, the result is seven different helical faces and is graphically represented on the helical net by performing a “one diagonal frame shift” for every rotation of the helix by one seven-residue diagonal. Each new face is designated sequentially from peptide-i to peptide-vii. It should be noted that there are only seven possible faces in an α-helix, i.e. if an 8th one diagonal frame shift is performed, the result is a face that is equivalent to peptide-i. Additionally, to prevent confusion regarding the terminology “a seven-residue diagonal,” the number “7” in the term relates to the number of residues displaced from residue number 1, in order to obtain residue number 2, each in their own seven-residue diagonal. Thus, the “7” in the term does not represent the number of amino acids in the resulting helical face. All the positive charges on the helical net plot are represented as dark blue (x, y) point markers. For the helical net of mPEP35-i, it should be noted that the residues within the solid black lines represent a helical face and therefore the residues to the left of the left-handed line and the residues to the right of the right-handed line are in another face, or more specifically, a multitude of other faces which are defined by mPEP35-ii to mPEP35-vii.
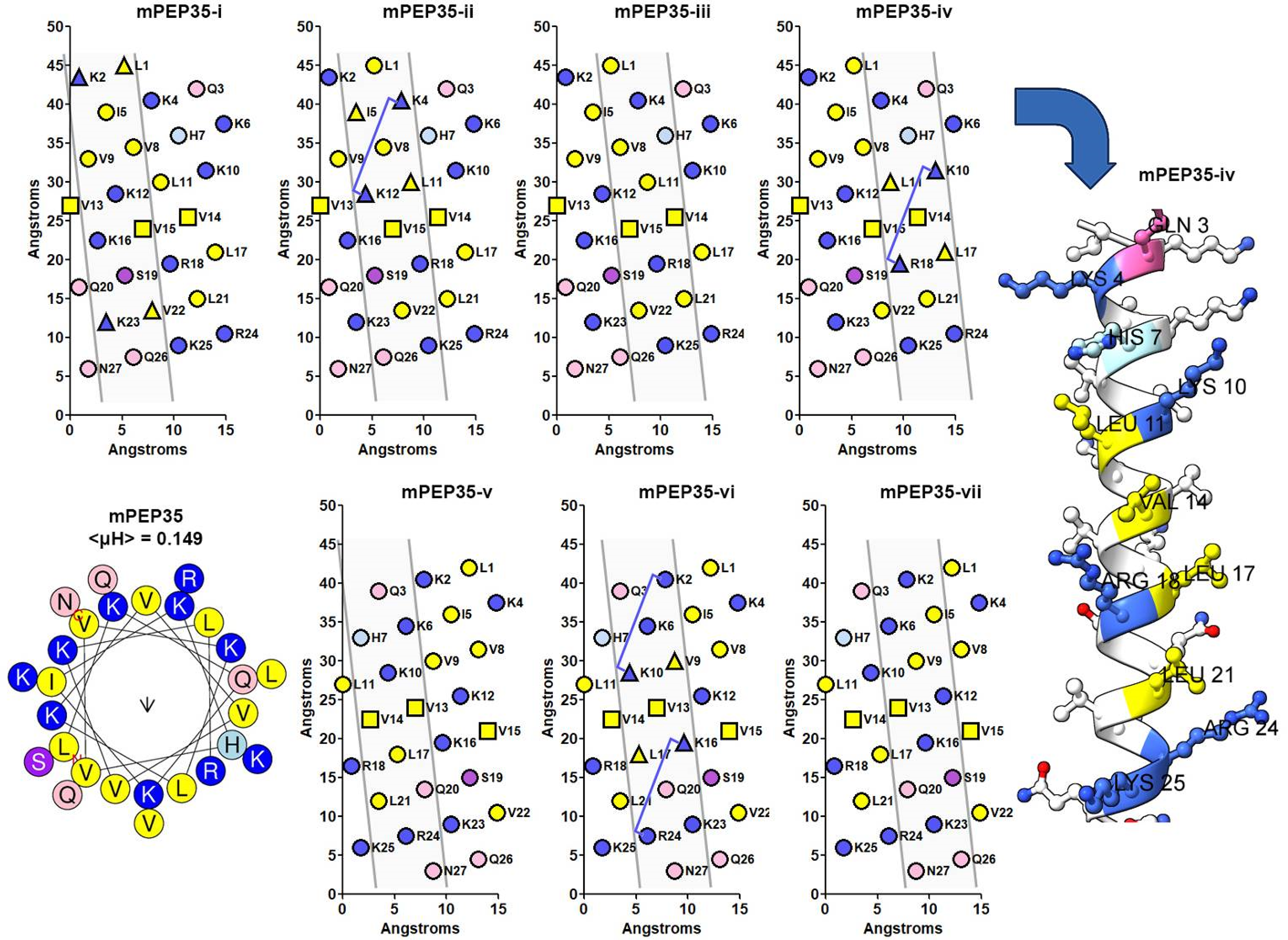
Multifaceted analysis of HA target peptide mPEP35. The helical nets depict the seven helical faces composed of three 7-residue diagonals with a representative model of the fourth face mPEP35-iv and a helical wheel. The four B1(X7)B2 HA binding domains are contained in three of the seven helical faces, i.e. mPEP35-ii, mPEP25-iv, mPEP35-vi (which contains two B1(X7)B2 HA binding domains). Positive residues in the helical face are colored in blue, hydrophobic in yellow, hydroxy-amino acids in purple and amide amino acids in pink. In mPEP35-i, the triangular shape for K23 and V22 in mPEP35-i is equivalent to Ziebell and Prestwich's residues K560 and V559 in RHAMM-binding domain II and K2 and L1 represent a similar orientation, 13 i.e. a positive/hydrophobic pair separated by one 7–residue diagonal. All other blue and yellow triangular pairs represent the same positive/hydrophobic pair. The B1(X7)B2 HA-binding domains are designated by a “tie-line” between two residues that follow the formula: B1 + 8 = B2. The three boxes (Val) is the tri-Val linker between RHAMM-binding domain I and II as per Zaleski et al. 4
This is the scenario exhibited by the positive control peptide mPEP35 and four target HA-binding peptides: 17x-3, #4, BHP3, and BHP4. In these cases, it is more appropriate to examine all the possible helical faces around the surface of the α-helix as defined by three adjacent seven-residue diagonals.
Concerning mPEP35, Figure 4, this peptide contains four B1(X7)B2 HA-binding domains, and furthermore, these four domains are presented by the α-helix in three helical faces, specifically, mPEP35-ii, mPE35-iv, and mPEP35-vi (which contains two B1(X7)B2 HA-binding domains). Additionally, in helical face mPEP35-i, residues K23 and V22 are indicated as triangles. This shape defines a region in Ziebell and Prestwich's work 13 that contains both a positively charged residue and a hydrophobic residue. The authors suggest that the hydrophobic residue adds stability to the RHAMM/HA-binding complex. Thus, the remainder of the seven helical faces of mPEP35 were searched for such a particular arrangement of amino acids. From Figure 4, it can be seen that helical faces mPEP35-ii, mPEP35-iv, and mPEP35-vi contain not only B1(X7)B2 HA-binding domains but also that particular arrangement of amino acids (denoted by a triangular shaped point marker) and it is assumed that this arrangement of amino acids will add stability to the peptide/HA-binding complex in these three helical faces. Lastly, the amino acids depicted on the helical net as square point markers represent the tri-Val linker that was used to covalently link RHAMM-binding domain I to RHAMM-binding domain II in the design methodology of mPEP35 4 ; thus, L1 to K12 define RHAMM-binding domain I and residues K16 to N27 define RHAMM-binding domain II.
Extension to the analysis and design of HA-binding peptide 17x-3
This is also demonstrated in Figure 5 for peptide 17x-3. In contrast to mPEP35 (which has four B1(X7)B2 HA-binding domains), 17x-3 contains five B1(X7)B2 domains in five of the seven helical faces, namely, 17x-3-i, 17x-ii, 17x-3-iv, 17x-3-v, and 17x-3-vii. Thus, not only does the 17x-3 design constitute a component of multivalency (i.e. it has one more HA-binding domain compared to mPEP35), but it confers a degree of multifacetedness compared to the mPEP35 design. Due to the distribution of HA-binding domains in 17x-3, this peptide has a 5/7 chance of presenting an HA-binding domain that can interact with a molecular interface such as HA compared to peptide mPEP35, which has only a 3/7 chance of presenting a HA-binding domain to HA. This may be a potential reason for the high degree of binding to HA demonstrated by 17x-3 compared to mPEP35 in the binding assay (Figure 3 of Koss et al., 2023). 16

Multifaceted analysis of HA target peptide 17x-3 16 with a representative model of the fourth face 17x-3-iv and a helical wheel. The helical nets depict the seven helical faces composed of three 7-residue diagonals. The five B1(X7)B2 HA-binding domains are contained in five of the seven helical faces, i.e. 17x-3-i, 17x-ii, 17x-3-iv, 17x-3-v, and 17x-3-vii. Amino acids, B1(X7)B2, and positive/hydrophobic pair designations as described in Figure 4. Residue color code is described in Figure 4.
Extension to the analysis and design of HA-binding peptide #4
Peptide #4 was also analyzed by this multifaceted characteristic of an α-helix (Figure 6). It should be first noted that peptide 17x-3 was designed in a similar manner to peptide mPEP35, i.e. mPEP35 has two contiguous (K2→K10, K10→R18) and two noncontiguous (K4→K12 and K16→R24) HA-binding domains, where contiguous is defined as two HA-binding domains sharing the same basic residue (the underlined residues in the above description). Thus, in the design of peptide 17x-3, this same feature was retained when plotting the helical net, as previously described. 16 This same configuration of three contiguous HA-binding domains is maintained in the design of peptide #4. However, the two noncontiguous domains were changed from noncontiguous to contiguous domains in the design of peptide #4. The net result for peptide #4 was five HA-binding domains contained within two sets of contiguous domains, i.e. K4→K12, K12→K20) and (K2→K10, K10→R18, R18→K26). This seemingly minor alteration to the design has a profound effect on the multifaceted analysis of peptide #4. Referring to Figure 6, the five B1(X7)B2 HA-binding domains are contained in four of the seven possible helical faces, i.e. 4-ii, 4-iv, 4-v, 4-vii (contains two HA-binding domains) in contrast to five HA-binding domains in 17x-3 being distributed in five of the seven helical faces. This effect is similar to that observed for the multifaceted analysis of mPEP35, where four HA-binding domains are distributed in three of the seven helical faces. Correspondingly, mPEP35 demonstrated a much lower level of binding to HA in comparison to 17x-3. A similar effect was observed for peptide #4 in the binding assay; #4 bound less to HA than that of 17x-3. 16 If there is a biophysical connection between the HA binding to the respective peptides and the distribution of HA-binding domains in the seven helical faces, this may explain the different results obtained for the binding of peptide #4 to HA relative to 17x-3.

Multifaceted analysis of HA target peptide #4. 16 The seven helical nets depict the seven helical faces composed of three 7-residue diagonals. The five B1(X7)B2 HA-binding domains are contained in four of the seven helical faces, i.e. 4-ii, 4-iv, 4-v, 4-vii (two HA-binding domains). Residue color code is described in Figure 4.
Extension to the analysis and design of HA-binding peptides BHP4
BHP4 is a variant of the peptide BH-P 21 and contains four HA-binding domains as two groups of contiguous domains, specifically (R4→R12, R12→R20) and (K6→R14, R14→K22). From the multifaceted analysis of BHP4 (Figure 7), the four HA-binding domains are contained within four helical faces. This is similar to the scenario of 17x-3, i.e. five HA-binding domains in five helical faces. This might be expected from the comparison of HA binding of 17x-3 (five domains in five faces) to peptide #4 (five domains in four faces), where peptide #4 has a diminished binding relative to 17x-3; BHP4 has similar binding to HA relative to peptide #4. This suggests that a reduced number of helical faces containing HA-binding domains might correlate with a reduced binding to HA. It should be noted that another research group also uses a similar set of peptide sequences, relative to BH-P, from human brain hyaluronan binding protein, which is designated b-HABP. 22
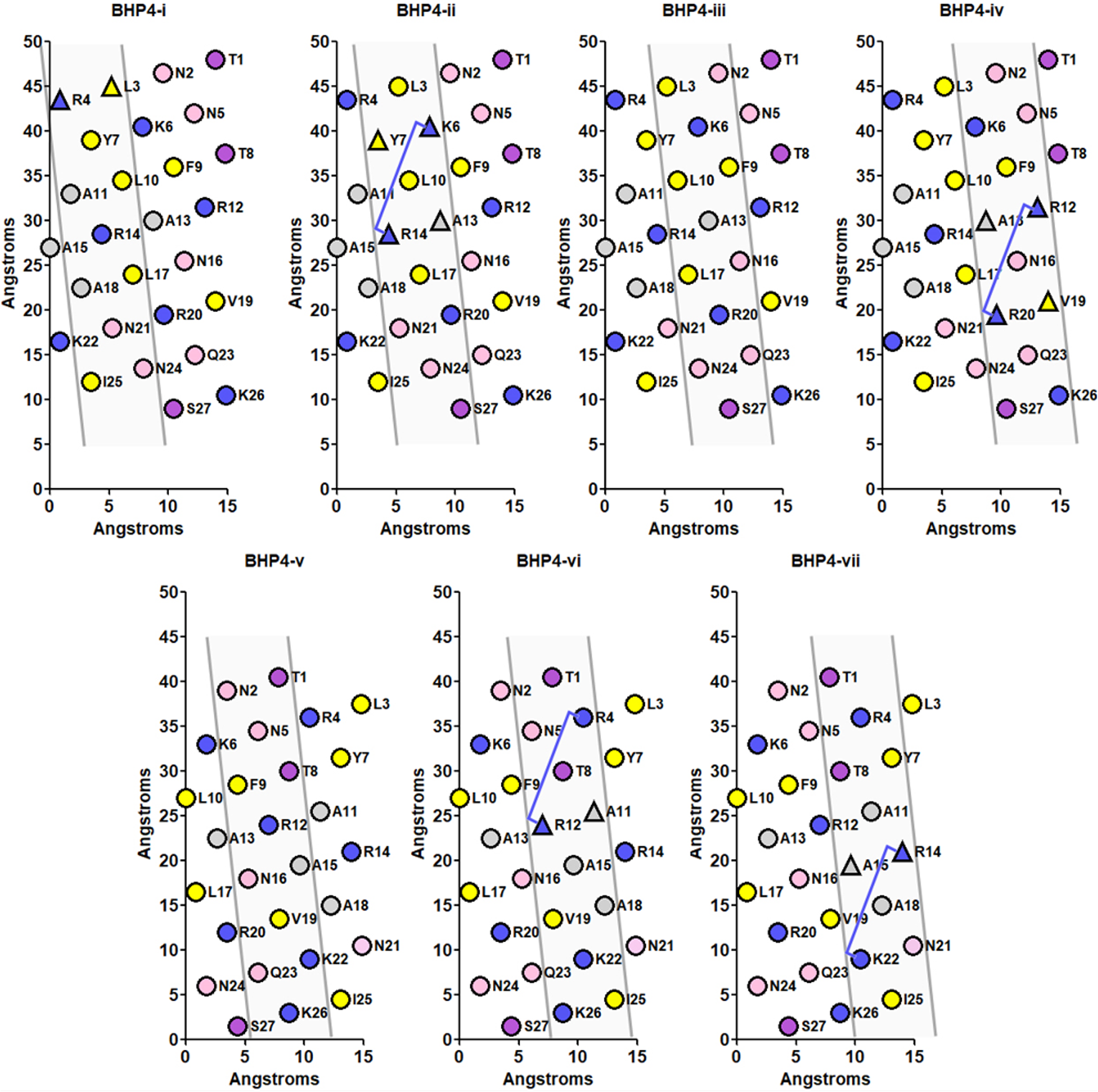
Multifaceted analysis of HA target peptide BHP4. 16 The seven helical nets depict the seven helical faces composed of three 7-residue diagonals. The four B1(X7)B2 HA binding domains are contained in four of the seven helical faces, i.e. BHP4-ii, BHP4-iv, BHP4-vi, BHP4-vii. Residue color code is described in Figure 4.
Additional considerations for the design of HA-binding peptides
With respect to HA-binding domains, i.e. B1(X7)B2, it has been suggested that positive charges in the center of the X7 region (position #4) adjacent to the B2 residue (position #7) or clustered within the X7 region will enhance the binding of a peptide to HA. 11 This claim was based on a model peptide R(G3)R(G)2R2, which demonstrated binding to HA similar to RHAMM-binding domains I or II. It should be noted that the sequence can be re-written, using the following notation: positionXregion 7 and explicitly written as: R1(GGG[4R]GG[7R])R2 to be consistent with our B1(X7)B2 designation (Note: The indicated formula is the basic mathematical expression, although in a helical peptide, the “position” in the notation positionXregion 7 will depend on the length of the peptide and the position of the B1(X7)B2 domain in the helix; thus, for a 27 residue peptide, the “position” can be any number from 2 to 26). Thus, a positive charge in the center of the X7 region, which we will designate as [4R7], or flanking the B2 residue, which we will designate as [7R7], should enhance binding to HA. mPEP35 and our target peptides 17x-3, #4 and BHP4 were analysed for these characteristics. mPEP35 (Figure 4) has one such [4R7] central positive residue in mPEP35-vi (specifically 6K7). Peptide 17x-3 (Figure 5) has one B1(X7)B2 domain that contains one [4R7] central residue in the 17x-3-ii helical face [15K7], peptide #4 (Figure 6) has two [4R7] central residues located in the #4-iv [6K7] and #4-v [16K7] helical faces, whereas, BHP4 (Figure 7) does not contain any [4R7] central residues. The binding of these peptides to HA are ordered in the following manner: 17x-3 > #4 > BHP4 > mPEP35, and the presence of a centrally located positive residue at [4R7] may be a contributing factor to the binding to HA.
With respect to the [7R7] residue, none of the peptides 17x-3, #4, or BHP4 have a positively charged residue at that position in the helical face containing a B1(X7)B2 domain. Additionally, with respect to the clustering of positive residues within the X7 region, peptides 17x-3, #4, and BHP4 were examined for this feature. For these three peptides, all were found to have “clustered” positive residues in the X7 region: 17x-3-i, ii, iv, v and vii; peptide #4-ii, iv, v and vii and BHP4-ii, iv, vi and vii; although, none of those positive charges were in the helical face containing the B1(X7)B2 HA-binding domains. Thus, these “clustered” positive residues might not contribute to binding to HA when HA is specifically bound to B1 and B2 residues of the B1(X7)B2-binding domain on the helical face. This doesn’t mean that these “clustered” positive charges do not contribute to HA binding, specifically in the case where there may be fiber formation, as demonstrated by transmission electron microscopy of the target peptides. 16 It should be noted that for every three 7-residue diagonal that defines a helical face, only five residues of the B1(X7)B2-binding domain will be presented on a helical face at any one point in time; those five residues being B1(1X7, 4X7, 7X7)B2 (in the basic mathematical expression). This can be seen in Figure 5, 17x-3-i, where the B1(X7)B2-binding domain is described as: K1(2T7, 3K7, 4A7, 5T7, 6V7, 7L7, 8I7)K9; although, only residues K1(2T7, 5T7, 8I7)K9 will be within the solid lines defining the 17x-3-i helical face.
Lastly, Ziebell and Prestwich 13 suggest that residues K548, K553 (corresponding to K16 in mPEP35), and K560 (corresponding to K23 in mPEP35) form electrostatic interactions with carboxylic acid groups in the GlcUA groups in HA and contribute to most of the stability to the RHAMM/HA complex, whereas, residues V538 (V9 in mPEP35) and V559 (V22 in mPEP35), interact with hydrophobic regions of GlcNAc. The authors suggest that the methylene groups of the sugar ring [i.e. –C(H2)–] are in close proximity to V538 and V559, adding some stability to the RHAMM/HA complex through hydrophobic interactions (due to the close proximity which could potentially exclude water). It has been suggested that carbohydrates are hydrophilic (due to the hydroxyl groups). However, they also exhibit some degree of hydrophobicity due to the methine groups, and the hydrophobicity is greatest when the methine groups are in the axial position of the “chair” conformation of the sugar ring,23,24 which generates a hydrophobic molecular interface. Ewurum et al. 25 provide a good visual depiction of the “hydrophobic face” in HA. This is in line with a review that indicates that HA is in a chair conformation and is also highly flexible, 14 and this flexibility might have an averaging effect on the hydrophobic interface presented by HA. It has also been suggested that the hydrophobic molecular interfaces in HA interact predominantly with aromatic residues in peptide/protein, e.g. Phe and Trp.24,26 Hudson et al. additionally indicates that aromatic amino acids predominantly interact with the methine groups of HA, although, through an electrostatic and electronic character (electropositive C-H bonds, i.e. partial positive charges on the C-H protons known as “electronic effects”) and furthermore suggests that hydrophobic interactions by themselves do not play a major role in binding. 27 Regardless of the mechanism of interaction, we consider the methylene or methine groups in HA to constitute a molecular interface that can interact with aromatic or aliphatic amino acids in proteins/peptides.
We examined the control peptide mPEP35 and the three HA-binding target peptides for hydrophobic or aromatic residue within those helical faces that contain an HA-binding domain. The multifaceted analysis of mPEP35 is depicted in Figure 4. Firstly, in helical face mPEP35-i, residues K23 and V22 are depicted as triangular shaped point markers, representing K560 and V559 in Ziebell and Prestwich. 13 It should be noted that each of these two residues are separated by an adjacent helical diagonal. Thus, a positively charged residue and a hydrophobic residue in this type of arrangement was searched for in the helical faces that contained a B1(X7)B2 HA-binding domain. For mPEP35, three helical faces that contained a B1(X7)B2-binding domain, each of the helical faces were shown to exhibit this arrangement, specifically, mPEP35-ii, mPEP35-iv, and mPEP35-vi. The hydrophobic residue in this orientation may add some stability to the peptide/HA complex. It is further noted that for the three helical faces in mPEP35, each face contains two such orientations of residues. Examination of 17x-3 (Figure 5) revealed that only three of the five helical faces contained such a positively charged residue/hydrophobic residue orientation. A similar scenario was observed for peptide #4 (Figure 6), i.e. for three of the four helical faces that contained a B1(X7)B2-binding domain, each of those helical faces contain only one of the positively charged residue/hydrophobic residue orientation. In contrast to 17x-3 and peptide #4, BHP4 (Figure 7), which contains four helical faces that contain B1(X7)B2-binding domains, has two faces that contain such an orientation. From the binding assay, 17x-3, peptide #4 and BHP4 demonstrated high binding to HA, suggesting that these orientations do not make a major contribution to HA binding.
Characterization of helical faces that do not contain B1(X7)B2-binding domain
From the HA-binding assay, 16 the plot demonstrates a decrease in specificity, in the order 17x-3 (five HA domains/five helical faces) > peptide #4 (five HA domains/four helical faces) > BHP4 (four HA domains/four helical faces) > mPEP35 (four HA domains/three helical faces).
A reduced number of helical faces containing HA-binding domains concomitantly results in an increased number of helical faces that do not contain HA-binding domains, thus presenting as a varied range of faces with various hydrophobic/hydrophilic characters, i.e. peptide molecular interfaces that may act as ligands which may be targets for other ECMs and/or other targets containing a corresponding interface. Table 1 demonstrates that fact. With respect to 17x-3 and #4, 17x-3 demonstrates a lower degree of variability relative to #4, i.e. 17x-3 contains five faces that are of medium hydrophobicity and two faces that are of low hydrophobicity. Peptide #4 is similar to 17x-3 considering that peptide #4 has four faces that are in the medium range and two that are in the low range of hydrophobicity, but is different from 17x-3 in that it contains a face that is in the high range of hydrophobicity. This potentially means that face #4-i may present a face that is capable of binding to another predominant hydrophobic molecular interface. There is a loss in specificity in peptide #4 relative to 17x-3 in that peptide #4 additionally binds to elastin, as well as HA. From Table 1 note 3, elastin is a protein that contains at least five segments that are possible hydrophobic molecular interfaces that have an < H > that ranges from 304 to 428. Thus, it is not surprising that peptide #4 additionally binds to elastin, as well as HA.
Hydrophobicity, <H>, for each of the seven helical faces.

The ranges termed “high,” “medium,” and “low” were determined by the absolute value of the lowest score, i.e. │-160│ = 160 and the highest score is 776, thus a total of 160 + 776 = 936. Divided by three ranges: 936/3 = 312. Thus, “high” = 465 + 312 = 777. The “medium” is = 464–312 = 152. The “low” range is 151–312 = −161.
The value of [488] is in the [high] range.
Elastin is a 786 amino acid residue protein (UniProt P15502, ELN, Human) which contains six regions that contain hydrophobic sequences that are between five and ten residues long, designated as follows: residue # to residue #, (sequence), e.g. (i) 253–261, A253AAAAAAKA261,<H> = 305 (ii) 302–315, A302AAAAAAAAAKAAK315,<H> = 428, (iii) 443–452 A443AAAAKAAKY452, <H> = 304, (iv) 489–498, A489AAAAKAAKAA498, <H> = 323, (v) 554–567, A554AAAKSAAKVAAK567, <H> = 358. 28
The type II collagen molecule has three identical α1-polypeptide chains. 29
Collagen alpha-1(II) chain (UniProtKB P02458, CO2A1, human). Hydroxy-Pro (symbol Hyp or O). There is only one Hyp in the α1-polypeptide chains. 30 There is no value for Pro in the scale (Figure 3), but Pro elutes at a relative retention time (relative to Gly) that is equivalent to Asn, thus, assigned Pro ∼ Asn.
The collagen type IIα1-segments: (i) A353GPPGPVGPAG363, <H> = 46, (ii) G372AKGEAGPTGARGPEGA388, <H> = 40.
Telopeptide of CO2A1: (i) N-telo peptide Q8TLVLLTLLVAA, 20 <H> = 735, (ii) C-telo peptide
I1462IDIAPMDIGGPEQEFGVD1480, <H> = 65.
Geltrex™ is a soluble form of basement membrane that contains: (i) laminin, (ii) collagen IV, (iii) entactin and (iv) heparan sulfate proteoglycans.
δ-toxin: MAADIISTIGDLVKWIIDTVNKFKK, <μH> = 0.583 31 .
PSMα3: MEFVAKLFKFFKDLLGKFLGNN, <μH> = 0.563 3 .
SynC sequence = MLSSLLRLRSLSLLRLRL, <μH> = 0.148. 32
With respect to BHP4, a similar pattern is observed. BHP4-ii, BHP4-iii, BHP4-vi, and BHP4-vii helical faces have hydrophobicity in the medium range, although three of those faces in BHP are slightly less hydrophobic than the corresponding medium hydrophobic faces in peptide #4. BHP4-i has a hydrophobicity value of 451 and this value is very close to the high range, i.e. 465–451 = 14 and 14/465 = 3% difference from the high range (or 7.5% difference from peptide #4-i). Thus, one could consider BHP4-i to be in the high range, which is similar to peptide #4. Additionally, BHP4-iv and BHP4-v are in the low hydrophobicity range, similar to peptide #4; although, those two faces, BHP4-iv and BHP4-v, are slightly less hydrophobic than peptide #4-iv and #4-v. It is not surprising that BHP4 has a lower specificity than peptide #4, i.e. BHP4-i is similar in hydrophobicity to peptide #4-i (suggesting a reason for BHP4 binding to elastin). Furthermore, the medium and low hydrophobicity faces of BHP4 are slightly less hydrophobic than those in peptide #4, and consequently, those faces will have very different binding characteristics relative to the corresponding helical faces in peptide #4. They may present as a more varied array of molecular interfaces that can interact with a more diverse set of peptides/proteins. This may be the reason for the loss in specificity for BHP4 relative to peptide #4, i.e. not only does BHP4 bind to HA, but to elastin (as in peptide #4) as well as collagen type II and IV and Geltrex (which contains basement membrane components like laminin, collagen IV enactin (also known as Nidogen). As noted in Table 1, footnote 4, two segments of collagen type II α1-polypeptide chains have an < H > value of 46 and 40. Collagen is composed of a GXaaYaa repeat (Gly, Xaa, Yaa signify Gly followed by any two amino acid residues), thus, the above indicated < H > is typical for segments within the helical region of collagen. BHP4-iv and BHP4-v have an < H > of 44 and 89, respectively. As well, mPEP35-vi and mPEP35-vii have an < H > of 72 and 83, respectively. Additionally, the N-telo peptide of collagen type II1α has an < H> = 735, and the C-telo peptide of collagen type IIα1 has an < H> = 65, Table 1, note 4.
Thus, both of these factors taken together, i.e. the decrease in number of non-B1(X7)B2 HA-binding faces and the variability in the hydrophobicity of each of the helical faces may account for the decrease in specificity observed, i.e. 17x-3 > peptide #4 > BHP4 > mPEP35.
Lastly, two peptides have been added to the bottom of Table 1 in order to demonstrate the difference between peptides of low amphipathicity (mPEP35 and the four target peptides) compared to peptides with a high degree of amphipathicity, namely two phenol-soluble modulins from bacteria that have high cytotoxicity (i.e. will lyse eukaryotic cells). Both δ-toxin (PSMγ) 33 and PSMα3 3 have three helical faces that are very hydrophobic, i.e. an < H > value of >600 and <μH> = 0.583 and 0.563 respectively.
The biological significance of the previous discussion is described herein. The extracellular matrix (ECM) is composed of a normal array of macromolecules that perform various functions, although it may be composed of two additional and specific types of molecular interfaces. Those interfaces are termed damage-associated molecular patterns (DAMPs) 34 and pathogen-associated molecular patterns (PAMPs). 35 Those molecular interfaces are termed endogenous or exogenous ligands, respectively and are generated by two different physiological processes of cell death, namely: (i) apoptosis (programed cell death, which is a normal process during regeneration of tissue) and (ii) necroptosis or alternatively, necrosis (triggered by injury or infection) + apoptosis = necroptosis (thus a post necrosis apoptosis).36,37
Both types of cell death result in distinct sets of cellular proteins released by these physiological processes. On the order of ∼2000 proteins were identified by liquid chromatographic mass spectroscopy (LC/MS/MS) in both apoptosis and necroptosis supernatants of dying cells. 37 Furthermore, Yu 38 suggests cellular components that are putative endogenous ligands include peptides, proteins, polysaccharides, proteoglycans, and nucleic acids and are ECM degradation products or arise from damaged tissue or dead cells. Knowing that there are ∼2000 proteins released in apoptosis or necroptosis, this is a substantial number of potential ligands that might be able to interact with the helical faces described by the multifaceted analysis of the indicated peptides, e.g. mPEP35. There are six known endogenous molecules that are released due to injury, including HA, substance P (an 11-residue peptide, RPKPQQFFLM), β-defensin, high-mobility group box 1, and heat shock protein-12. 39 Furthermore, through the normal process of catabolism, intracellular proteins are recycled through the action of proteasomes, which enzymatically degrade proteins into peptides, and finally, amino acids. 40 In terms of exogenous ligands, macromolecules that are on the surface of microbes, 41 e.g. bacteria, viruses, and fungi, 35 are composed of molecular patterns, which are very specific to their source and thus, highly variable. These macromolecules may present as a varied degree of hydrophobicity/hydrophilicity and charge distribution in a similar fashion to the scenario we are seeing in the multifaceted analysis of our target helical peptides.
Application of methodology to the characterization of other α-helical peptides
In order to determine the applicability of the multifaceted analysis of peptides other than those containing B1(X7)B2 domains, another peptide was selected for analysis by our methodology, namely a synthetic modification to cytochrome oxidase subunit IV (Syn C or SynC—P04037 COX4_YEAST). 32
SynC is a synthetic mitochondrial presequence.42,43 Gavel et al. (1988) searched segments of the peptide for regions of maximum hydrophobicity and, furthermore, incorporated a methodology to account for degrees of freedom of rotational isomers (essentially rotamers)44,45 for each of the amino acids. Subsequently, the authors developed a “modified” helical wheel, which resulted in the amino acids being grouped into four sectors around the helical wheel diagram (Figure 8). SynC has a hydrophobic moment of 0.148, as per HeliQuest. In order to put this <µH > into perspective, SynC can be ordered as follows (from Figure 1): cell-penetrating peptide (0.570) > RHAMM-binding domain I (0.453) > AX9, where X = Ala (0.371) > BHP3 (0.234) > 17 × 3 (0.199) > SynC (0.148) > peptide #4 (0.072) > naA (0.010). Thus, from this perspective, one would assume that SynC has a low degree of amphipathicity; although, in Gavel et al.'s work, their “modified” helical wheel yields a hydrophobic moment (µH, as per Eisenberg, 1982, equation (1)) that is 2.3-fold higher (6.0/2.6 = 2.3) than that calculated using the “classical” helical wheel. 46 Thus, the classical helical wheel only depicts the position of the α-carbon atom on the wheel for each of the amino acid residues and does not take into account the rotational degrees of freedom for each of the side chain atoms.

Multifaceted analysis of mitochondrial targeting peptide SynC 32 with a representative model of the fourth face SynC-iv and a helical wheel. The helical nets depict the seven helical faces composed of three 7-residue diagonals. The top-down view depicted by Gavel et al. denotes SynC faces i, ii, iv, vi/vii in four quadrants with amino acids plotted in a counter-clockwise orientation, as opposed the clockwise shown in a helical wheel. The seven helical faces, i.e. SynC-i-vii are presented in a net form to display the face-by-face amphiphilicity. Residue color code is described in Figure 4. Copyright obtained from John Wiley and Sons.
We compared our multifaceted analysis of SynC to the results obtained by Gavel et al. From Table 1, it is observed that all of the helical faces SynC comprise hydrophobicity values that fall into the “medium” range as designated by the criteria described by Table 1, footnote 1; of those six faces, four of them are very close to the high hydrophobicity range, i.e. 77–80% of the high range. Thus, one might expect those three helical faces to be significantly hydrophobic as well. The multifaceted analysis of SynC thus suggests that there are indeed very hydrophobic faces in SynC which may not be apparent from the <µH > value and furthermore suggests a similar conclusion as obtained by Gavel et al. A detailed analysis of this comparison is reported in Figure 8 and Table 2. From Figure 8, it can be seen that the helical wheel (note the direction of the helical wheel is counter-clockwise, relative to the HeliQuest helical wheel) can be divided into four major sectors as defined by the “modified” helical wheel of Gavel; et al., i.e. SynC-i, SynC-ii, SynC-iv, and SynC-vi/vii. From the helical wheel, it can be seen that the sectors are composed of amino acids that are “neighboring” amino acids, with respect to the helical wheel designation, and are in groups of four or five residues. This is not surprising based on the helical wheel of AX9, where the hydrophobic face is composed of seven “neighboring” Ala residues. For clarity, SynC-iii and SynC-v contain sectors a/b and predominantly c, respectively. Thus, those sectors in Gavel 32 are essentially “minimal” helical faces based on AX9. Since Gavel applied a “rotamer” analysis to their design, it is apparent that one would obtain a tightly grouped set of amino acids in the designated sectors. This aspect highlights the utility of a “rotameric analysis” to applications of engineering design of peptides, especially when the peptide molecular interface or partner “binding pocket” is of interest in the design. Furthermore, from Table 2, the seven helical faces of SynC can be further divided into sectors a, b, a/b, c, c/d, and d sequentially. Lastly, it can be seen from Table 2 that the multifaceted analysis of SynC compares very well with that of the Gavel “modified” helical wheel, i.e. each one of the helical faces contains either four or five of the amino acids that are contained in sectors a, b, a/b, c, c/d and d sectors. Additionally, for the faces {i} to {vii}, for each of the amino acids that do not coincide with one of the sectors a, b, a/b, c, c/d, and d, those amino acids generally are in an adjacent sector. This fact is not surprising because in the generation of the seven helical faces {i} to {vii}, going from one face to the next, e.g. {i} → {ii}, involves a one 7-residue diagonal frame shift to generate face {ii} and subsequently helical face {ii} will contain at least two 7-residue diagonals from helical face{i}. The close alignment of the multifaceted analysis with the “modified” helical wheel of Gavel (1988) validates that our method is capable of detecting “hydrophobic character” in those peptides that do not demonstrate an apparent amphipathicity. From the previous discussion, it is apparent that the multifaceted analysis is a methodology that may be used for detecting hydrophobic faces or “hydrophobic molecular interfaces” in peptides that demonstrate a low <µH > .
Comparison of multifaceted analysis of SynC mitochondrial targeting peptide MLSSLLRLRSLSLLRLRL.

Lowercase letter in italics and
AA denotes amino acid in the single letter designation.
Discussion
A multifaceted approach is used to analyze modeled peptides for the varied characteristics of the faces presented by an amphipathic α-helical peptide, using a minimalistic engineering design, based on two series of peptide analogs. This approach demonstrated that one series of analogs (AX9) demonstrated a hydrophobic face, containing three 7-residue diagonals in a helical net, which constitutes a basic helical face of an α-helical peptide. A second series of analogs (AX7) demonstrated a hydrophilic face, that is complimentary to the hydrophobic face of the AX9 series of analogs (i.e. both hydrophobic and hydrophilic faces when combined would reproduce the entire surface of an α-helix). An α-helix contains seven distinct faces presented by a helical surface. The basic helical face (described by the AX9 series of analogs) was used to analyze HA-binding peptides that varied in number and composition of HA-binding domains and peptide <μH>, specifically 17x-3, peptide #4 and BHP4. It was demonstrated that the multifaceted analysis revealed that the strong HA binders (including high to moderate specificity) presented helical faces that were optimized designs relative to the positive control peptide mPEP35. More specifically, 17x-3 presented five HA-binding domains in five of the helical faces; whereas, mPEP35 presented four HA-binding domains in three helical faces. This suggests that the optimized versions of the HA-binding peptides were dependant on the distribution of the HA-binding domains, resulting in high binding and specificity. If this is correct, it may be possible to design peptides with up to seven binding domains distributed per seven faces, but this is not possible without extending the peptide length considerably. This is a limitation, as longer peptides will likely misfold in unpredictable ways, resulting in limited binding characteristics, and may not be possible to synthesize with conventional solid phase methods. These tailored designs could be optimized with computational techniques, but this is beyond the conceptual scope of this report.
In order to test the hypothesis, a biologically relevant peptide, SynC, was analyzed by the multifaceted analysis and it was shown that this analysis resulted in a highly similar result to that performed by Gavel et al. 32 These researchers used a rotameric analysis to group the amino acids presented on the helix and demonstrated that there were indeed at least three helical faces that presented very hydrophobic faces, even though the overall <μH > was small, i.e. (0.148). Our numerical and qualitative multifaceted analysis revealed a highly similar result, with amino acids being grouped as per Gavel et al. or were displaced by one helical face (a very reasonable result considering that the multifaceted analysis rotates by one diagonal at a time, indicating that two diagonals in the adjacent face come from the preceding helical face).
Understanding the content of binding domains and how they are arranged in helical faces can allow for the exploration of HA and other glycan fragment binding in inflammation. We further commented that since a reduced number of HA-binding faces concomitantly results in more faces that do not contain an HA-binding domain; thus, may present faces as varied as those found in DAMPs and PAMPs. The arrangement of hydrophobic residues can also have implications in designing potent AMPs. The Hodges’ group demonstrated efficient drug design in which they incorporated what they termed “specificity determinants,” i.e. incorporation of very specific amino acids into the hydrophobic face of AMPs in order to maintain antimicrobial activity, but reduce hemolytic activity toward eukaryotic cells. 47 In one first model, they incorporated two hydrophilic residues into the center of the hydrophobic face of an alpha-helix (termed specificity determinants) and in the second case, as well as including specificity determinants in the hydrophobic face, they incorporated Arg, Lys and analogous of Lys into the hydrophilic face. 47 The result demonstrated excellent antimicrobial activity and essentially no hemolytic activity. These works highlight the engineering philosophy of our analysis of helical faces in order to analyze and incorporate engineering design elements into helical peptides in order to achieve desired therapeutic drug candidates.
Conclusions
From the preceding discussion, designing synthetic peptides requires a multifaceted approach in order to incorporate parameters into the peptides that will result in optimal designs. Alpha helical peptides have seven distinct faces as provided by the example minimalistic peptide AX9 analogs, i.e. each face being composed of three 7-residue diagonals. This was evidenced by the drastic chromatographic retention time differences as the X9 residue was changed throughout the entire range of amino acid amphipathicity. This was contrasted by the hydrophilic AX7 face, which did not exhibit chromatographic separation as well as the hydrophobic face of the AX9 series of peptides to the C8 hydrophobic column interface. This was expanded to our family of helical HA-binding peptides and evaluated against their binding affinities. When it comes to peptides of very low amphipathicity, one must investigate the very nature of all of the helical faces in order to gain a better understanding of the potential characteristics of the peptide in its entirety, which cannot be assessed by a mere averaging effect that will be obtained from deriving the <μH > . Furthermore, the similarities between 17x-3 and peptide #4 in contrast to the similarities in mPEP35 and BHP4 demonstrated by the multifaceted analysis might reflect the specificity observed in prior binding data. In our future work, a machine learning program is being developed to elucidate binding criteria and novel HA-binding sequences, based on the differences as examined by the multifaceted analysis of the helical faces of these peptides. The focus of the analysis will be on (1) the arrangements of the B1(X7)B2-binding domains and (2) the hydrophobic to hydrophilic ratio of amino acids in order to assess the most proficient reported biofunctionalities, including HA-binding and mitochondrial delivery, which is impactful across a wide array of biotechnology and medical therapeutics.
Footnotes
Abbreviation Definition
Author contributions
TJS conceptualized the entire project, performed and oversaw the analytical work, generated the figures, wrote and edited the final document. JB performed the molecular simulations of each peptide and presented their faces according to the orientations of the helical nets and wheels. PS synthesized and performed the analytics on the peptides for this study. ASA and JAW oversaw and assisted in the conceptual design of this work. KMK worked with TJS to conceive this project, oversee recent elements of the work (modeling and data interpretation), design this manuscript and portray the helical nets/wheels, and generate the final manuscript. All authors assisted in editing and finalizing the manuscript for publication.
Conflict of interest
The authors of this work have nothing to disclose.
Funding
The authors of this work would like to thank their funding from the American Heart Association (AHA) 20POST35210774 and the Canadian Institutes of Health Research (CIHR) RN409371 - 430628. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.