
Abstract
Idiopathic pulmonary fibrosis is a chronic and progressive interstitial lung disease with a poor prognosis. Idiopathic pulmonary fibrosis is characterized by repeated alveolar epithelial damage leading to abnormal repair. The intercellular microenvironment is disturbed, leading to continuous activation of fibroblasts and myofibroblasts, deposition of extracellular matrix, and ultimately fibrosis. Moreover, pulmonary fibrosis was also found as a COVID-19 complication. Currently, two drugs, pirfenidone and nintedanib, are approved for clinical therapy worldwide. However, they can merely slow the disease's progression rather than rescue it. These two drugs have other limitations, such as lack of efficacy, adverse effects, and poor pharmacokinetics. Consequently, a growing number of molecular therapies have been actively developed. Treatment options for IPF are becoming increasingly available. This article reviews the research platform, including cell and animal models involved in molecular therapy studies of idiopathic pulmonary fibrosis as well as the promising therapeutic targets and their development progress during clinical trials. The former includes patient case/control studies, cell models, and animal models. The latter includes transforming growth factor-beta, vascular endothelial growth factor, platelet-derived growth factor, fibroblast growth factor, lysophosphatidic acid, interleukin-13, Rho-associated coiled-coil forming protein kinase family, and Janus kinases/signal transducers and activators of transcription pathway. We mainly focused on the therapeutic targets that have not only entered clinical trials but were publicly published with their clinical outcomes. Moreover, this work provides an outlook on some promising targets for further validation of their possibilities to cure the disease.
Therapeutic targets for molecular therapy of idiopathic pulmonary fibrosis.
General introduction of idiopathic pulmonary fibrosis (IPF)
Definition, characteristics, prevalence, and patient population of IPF
IPF is one of the most prevalent, progressive, chronic, and irremediable interstitial lung diseases (ILD) with unknown mechanisms. 1 IPF is characterized by the destruction of normal lung tissue, continuous generation and activation of myofibroblasts, accumulation of extracellular matrix, and eventually fibrosis of the lung. 2
The prognosis is indeed poor since the median survival is 2–5 years from the diagnosis.2,3 The number of patients is currently on the rise worldwide, possibly due to an aging population, increased awareness of the disease, etc. People suffering from IPF have an average onset age of 65–70 years, and the chance of getting the disease is proportionate to age. Furthermore, men are at greater risk of developing the disease. The onset and progression of IPF have also been implicated in the inhalation of particulate matter. A history of smoking correlates with the progression of the disease in most patients, and other environmental factors such as wood debris, agriculture, viruses, rocks, and silica have also been shown to be correlated with its development 2 (Figure 1).

The risk factors of idiopathic pulmonary fibrosis (IPF). It was created with Biorender.com.
There is variation in the reported incidence and prevalence of IPF worldwide due to different research methods.4,5 Some studies have reported that the incidence of IPF ranges between 2.8 and 18 cases for each hundred thousand people yearly in Europe and North America. The incidence in Asia and South America is likely lower, probably 0.5–4.2 cases for every hundred thousand people yearly. 2
Poor clinical outcome and difficulty in the clinical management of IPF
Although much progress has been made in the study of IPF, many problems remain. The first challenge is the clinician's diagnosis of IPF, whose symptoms are very similar to those of other interstitial lung diseases, and the clinician's decision is based solely on morphological criteria. 2 Secondly, the course of disease in patients with IPF is highly individual, some patients experience a highly variable and unpredictable course, and a sharp decline in lung function, known as acute deterioration, can occur at any point in the course of the disease. 5 Many patients start with reduced lung compliance and weakened gas exchange, gradually progressing to respiratory failure or even death. Now, only pirfenidone and nintedanib are officially used in clinical treatment. 6 However, these two drugs only slow the fibrosis progression and do not cure or reverse the disease. In addition, lung transplantation has been shown to improve patient survival significantly, but the therapy is only available for a few patients.
Present research methodology for IPF
Patient case/control studies
As mentioned above, the diagnosis of IPF patients is not easy, and the prognosis is poor, as well as the variability of disease progression varies from person to person, which has led researchers to explore biomarkers from patients’ fluid specimens that may be helpful for diagnosis and prognosis. Meanwhile, the analysis of certain genetic abnormalities in patients’ specimens can also contribute to the investigation of pathological mechanisms. Currently, proteins such as cytokines, and genetic mutations such as polymorphisms, in patients’bronchoalveolar lavage fluid, blood, or serum, are most commonly studied, and molecular research interest in sputum is also gradually increasing. 7 When studying abnormally expressed targets in specimens from patients with IPF, in addition to the most commonly used healthy controls, samples from patients with non-IPF chronic ILDs, such as sarcoidosis or myositis-associated ILD, can be added as disease controls to determine whether the biomarker is specific.8–10
Thanks to the cohort and case/control studies, many IPF-related biomarkers have been discovered. For example, the biomarkers reflecting alveolar epithelial dysfunction are surfactant proteins A and D (SP-A and SP-D), 8 Krebs von den lugen-6 (KL-6), 11 mucin 5B (MUC5B) gene polymorphism, 12 etc. The immune dysregulation-related biomarkers include chitinase-3-like protein 1 (YKL-40), 13 CC chemokine Ligand-18 (CCL-18), 14 and so on. The present biomarkers of extracellular matrix remodeling are matrix metalloproteinases (MMPs), such as MMP-1 and MMP-7, 15 periostin (POSTN), 16 lysyl oxidase-like (LOXL) proteins,7,10,17 etc. Although there have been a large number of biomarker studies, the performance of these identified biomarkers has not been fully validated yet.
Cell models
Damage and remodeling of the lung epithelium promote the development of IPF. During this process, fibroblasts are continuously activated to proliferate and differentiate into myofibroblasts, the primary effector cells of the fibrotic response, and therefore have become the major interest cells extensively studied by many scientists. In addition to the above important cellular involvement, endothelial cells, vascular smooth muscle cells, and immune cells such as T cells, macrophages, and neutrophils are also thought to contribute to IPF. 18 Patient tissue specimens for IPF were taken from puncture biopsies or pathological lung specimens that have undergone lung transplantation, 19 while control tissue was derived from normal lung tissue of organ donors, or surgical lung specimens from patients undergoing resection surgery.20,21 By contrast, the lungs from animal models were more available. In cell studies of IPF, most experiments are based on two-dimensional cultures on a plastic/glass interface. 22 The isolation and culture protocols of these cells are thus described below.
Lung fibroblasts
Fibroblasts are the core cells of the disease and are easy to be isolated and cultured. The human lung specimens were cut into pieces and cultured in a rapid-growing mycobacteria medium for a while to grow a monolayer of fibroblast-like cells before being digested with trypsin to obtain freeze-preservable primary cells, 20 and further routinely cultured. The isolation and purification of mouse fibroblasts were performed as previously described. 23 In brief, after obtaining intact mouse lungs, cut into pieces, digested with collagenase and DNase in Dulbecco's modified eagle medium (DMEM), filtered, centrifuged, and the resulting suspension was washed and cultured with the same daily protocol as human fibroblasts. 24 In addition, human normal lung fibroblast cell lines have also been used for pulmonary fibrosis studies, such as WI-38 cells, IMR-90 cells, and normal human lung fibroblast (NHLF) cells.25–27 WI-38 cells and IMR-90 cells were incubated in DMEM medium, while NHLF cells were incubated in FGM™-2 fibroblasts growth medium-2 BulletKit.
Bronchial epithelial cells
Primary human bronchial epithelial (HBE) cells were isolated from bronchi of excess pathologic tissue after lung transplantation or donated organs unsuitable for transplantation. Normal fresh isolated HBE cells were transferred onto rat-tail type I collagen-coated Petri dishes, cultured overnight, and finally changed into the bronchial epithelial growth medium (BEGM).28,29 The HBE cells were cultured in a completed BronchiaLife epithelial airway medium. 30 When mouse broncho-alveolar epithelial cells were extracted, flushing the lungs with saline through the pulmonary artery, injecting neutral protease into the lungs via the trachea, following the injection of low melting point agarose solution with crushed ice under the lungs to polymerize the agarose. Then, the lung tissue was incubated in sterile tubes and separated from the airway in a DMEM matrix containing DNase I, filtered, centrifuged, and resuspended in DMEM. The crude cell suspension was plated on anti-CD45 and anti-CD32 antibodies-coated dishes, and the unattached cells were collected after panning, and daily incubated in Ham's F12 medium. 31 The normal bronchial epithelial cell line BEAS-2B was incubated in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic (AA) solution, or BEGM supplemented with 1% AA.
Type Ⅱ alveolar epithelial cells
Human lung tissues were minced and digested with dispase II, elastase, and DNase I. Single cell suspension was prepared by using enzymatic digestion and validated by using flow cytometry for its specific markers such as CD31, CD45, and EpCAM. 32 The cell suspension of mouse alveolar type II epithelium was prepared like for mouse bronchial epithelial cells. In order to purify the cell suspension, it was treated with biotinylated anti-CD32 and anti-CD45 antibodies, centrifuged, resuspended in DMEM medium, incubated with magnetic pellets coated with streptavidin, and then aspirated from the bottom of the tube through a magnetic tube separator, centrifuged, recultured in DMEM and finally stained with Taipan Blue to determine cell viability. 33
Lung endothelial cells
Mouse lung endothelial cells were isolated by firstly cutting freshly isolated mouse lungs and then digesting them with collagenase type I for 45 min. The digest was ground with a 12 cm cannula and filtered through a 70-μm cell sieve, and the resulting filtrate was centrifuged at 300g for 8 min at 4 °C. The cells were resuspended in fresh M199 buffer and reacted for 4 h with immunomagnetic beads coupled to CD31 and CD144. The bound lung cells were then inoculated into collagen-coated tissue culture dishes and cultured for at least 3 days before further experiments. 34 Endothelial colony-forming cells (ECFCs) are a population of cells capable of differentiating into endothelial cells and forming new blood vessels in adults. They have been used in the study of several lung diseases, including IPF. ECFC was isolated from the whole peripheral blood of IPF patients and controls. Blood samples were diluted with PBS/EDTA and centrifuged at a 1200g density gradient for 20 min. The obtained mononuclear cells were washed twice with PBS/EDTA in a specific endothelial cell growth medium (EGM2) supplemented with 10% fetal calf serum (FCS), and the cells were inoculated in the medium at a density of 5 × 106 cells per well in six-well plates. The dishes were screened twice a week for ECFC growth. Each new colony was isolated and cultured. 35
Pulmonary artery smooth muscle cells (PASMCs)
During pulmonary fibrosis, not only is the lung parenchyma damaged, but the pulmonary vasculature is also affected, with microvascular abnormalities and macrovascular remodeling occurring. 36 Studies have shown that abnormal vascular remodeling is associated with the pathogenesis of IPF. 37 Mouse PASMC isolation was performed by digesting pulmonary artery tissue in a medium containing CaCl2 and elastase, collagenase, soybean trypsin inhibitor, and albumin for 20 min at 37 °C. After filtration with 100-μm cell filters, cells were incubated with Dynabeads coated with anti-cluster of differentiation (CD) 31 and anti-CD102 antibodies. The remaining SMCs were centrifuged at 225g for 6 min at 4 °C and incubated in DMEM antibiotic solution containing 10% FBS. To confirm the isolation of PASMCs, cells were validated by positive staining for α-smooth muscle actin with antibodies.38,39
Animal models
Animal models are critical for an in-depth study of disease progression. However, there is a lack of spontaneous experimental models for IPF, so it is essential to establish animal models mimicking the characteristics of the disease. Numerous animal models of pulmonary fibrosis have been developed by researchers, but the most commonly used and easily established one is the bleomycin-induced model.
Bleomycin (BLM)
BLM is capable of causing lung damage due to the lack of BLM hydrolase in the lungs. The mechanism of lung damage caused by BLM is mainly the production of superoxide and hydroxyl radicals that cleave DNA, leading to single or double-stranded DNA break.40,41 The most important advantage of the BLM model is that it reproduces the typical features of human IPF, which are very similar in some molecular histopathological characteristics, and the other advantage is that the BLM model is relatively simple to operate, easy to induce, and reproducible.41,42
The sensitivity to bleomycin varies between strains of mice, among which C57BL/6 is the most sensitive and responsive to the drug. The majority of research was conducted on male mice aged 8–12 weeks.40,43,44 There are various routes of administration, such as intraperitoneal, subcutaneous, and endotracheal, but the most common and best is the endotracheal BLM model, which allows the damage to be well confined to the lungs. 40 Intratracheal administration can be divided into oral drips and direct intratracheal injections. 42 Direct intratracheal injections require a tracheotomy book and are prone to cause animal death, while intratracheal drips are relatively safer. In terms of dosing frequency, although it has been suggested that repeated dosing can compensate for the self-limiting nature of fibrosis in mice by constructing persistent fibrosis, which means both increased time and cost, therefore a single administration at the optimal dose is preferred by investigators.
A frequently mentioned disadvantage of the BLM model is the self-limiting development of pulmonary fibrosis in young mice. 41 However, the course of human IPF is progressive. Although it is not commonly observed before the mice are sacrificed in regular models except for long-term observation, this remains a major concern for some researchers.
Fluorescein isothiocyanate (FITC)
FITC is a small fluorescent molecule and semi-antigen, which can cause acute lung injury after intratracheal administration in a week and develop into fibrosis within 2–3 weeks.45,46 Both BALB/c and C57BL/6 mice are susceptible and easily induced, 45 with Balb/c mice showing more significant fibrosis to a given dose of FITC. The most significant advantage of this FITC-induced model is that the fluorescent molecules deposited in the fibrotic lung tissue can be easily observed, allowing better tracking of the lesion site, in addition to the fact that the effect of FITC is sustained and long-term, maintaining fibrosis for at least 6 months without self-limiting regression. 45 The disadvantages include that the model lacks some of the typical pathological features of IPF, such as fibroblastic foci, and the FITC-induced fibrotic response exhibits high diversity among different batches, thus affecting the stability and reproducibility of the model.40,47
Silica
The silica-induced lung fibrosis model resembles the fibrous nodule damage that forms in humans after exposure to mineral fibers. 48 The routes of administration are nebulization, tracheal injection, or transoral endotracheal drip.40,49 Currently, the most popular approach is injecting silica suspension directly into the trachea through a tracheotomy. 49 The intratracheal model is relatively faster and easier to induce fibrosis within 2–4 weeks, whereas nebulized administration takes longer. Intratracheal injection of silica is more susceptible to induce fibrosis in C57BL/6 than in CBA/J mice. 50 Silicon dioxide fibrous nodules can be identified by histology and polarized light microscopy. 40 The advantages of this model are that silica is not easily cleared from the lungs, and the fibrotic response lasts longer. In addition, lung nodules at the site of silica deposition can be easily observed. The disadvantages are the lack of some of the histopathological features of IPF and low reproducibility for the timing of modeling.40,47
Radiation
In mice, a single total body radiation of 12–15 Gγ can induce pulmonary fibrosis, which is mainly dependent on inflammation and free radical-mediated DNA damage. 51 However, localized chest radiation is more commonly used and targeted. C57BL/6 mice are easier to induce than other strains. Pulmonary fibrosis is evident following 24 weeks after local radiation of 16 Gγ to the chest of C57BL/6. 45 Radiation-induced models can be used to investigate differences in susceptibility to IPF in inbred mice, but the time required for induction is long, and sophisticated radiation equipment is required, leading to a tight budget.40,45,47
Validation of lung fibrosis in the model is also a necessary and primary process. Masson trichrome histological analysis, HE staining, and Ashcroft scoring can quantify the extent of fibrosis, hydroxyproline measurements reflect collagen content, while analysis of cell types and numbers in bronchoalveolar lavage fluid (BALF) can provide insight into fibrosis markers and inflammation intensity. In the meantime, α-smooth muscle actin measurements can be used to assess the differentiation of myofibroblasts.40,42,49,52–54
Presently reported therapeutic targets in IPF
Using some of the models mentioned above, researchers have not only resolved the molecular mechanisms of disease development to a certain extent, but also obtained some targets with therapeutic value, and some of them have been developed into drugs for clinical application. Up to time now, the specific pathogenesis of IPF has yet to be fully elucidated, but great progress has been made in further understanding the disease. The most widely accepted theory is that the interactions with mesenchymal cells, endothelial cells, and immune cells following repetitive alveolar epithelial cell injury dysregulation trigger an aberrant wound healing response and finally pulmonary fibrosis. 55 During this process, abnormally activated cells secrete a large number of cytokines, chemokines, and growth factors, accompanied by aberrant activation of many signaling pathways.
Some of these mediators and their associated signaling pathways have become the focus of current research while exploring candidate therapeutic targets. Pirfenidone and nintedanib are the only two drugs in clinical use in some countries and regions. Pirfenidone is an oral drug with some side effects, mainly gastrointestinal and cutaneous reactions, which are manageable in most patients. It acts to inhibit fibrotic, inflammatory, and oxidative responses through down-regulation of transforming growth factor-β (TGF-β) signaling, thereby reducing the cellular synthesis of collagen and related fibronectin, as well as suppressing the phosphorylation of its signaling pathway molecules such as Smad3 and p38, inhibiting the release of inflammatory factors and reducing oxidative stress.6,56,57 Nintedanib is also used orally, and some patients experience diarrhea, but still tolerable for the majority of patients. Nintedanib treatment can down-regulate fibroblast proliferation and migratory activity, and inhibit TGF-β-induced myofibroblast transformation.1,58,59 It is worth noting that the current drug regimens for pulmonary fibrosis associated with neocoronary pneumonia have been developed mainly around the relevant guidelines for IPF. These two anti-fibrotic agents have emerged as potential treatment options for post-neoconiosis pulmonary fibrosis.60,61 Some important molecular targets and their progress in current research are discussed in detail (Table 1).
Molecular drugs that are currently in or have completed clinical trials and their corresponding information.
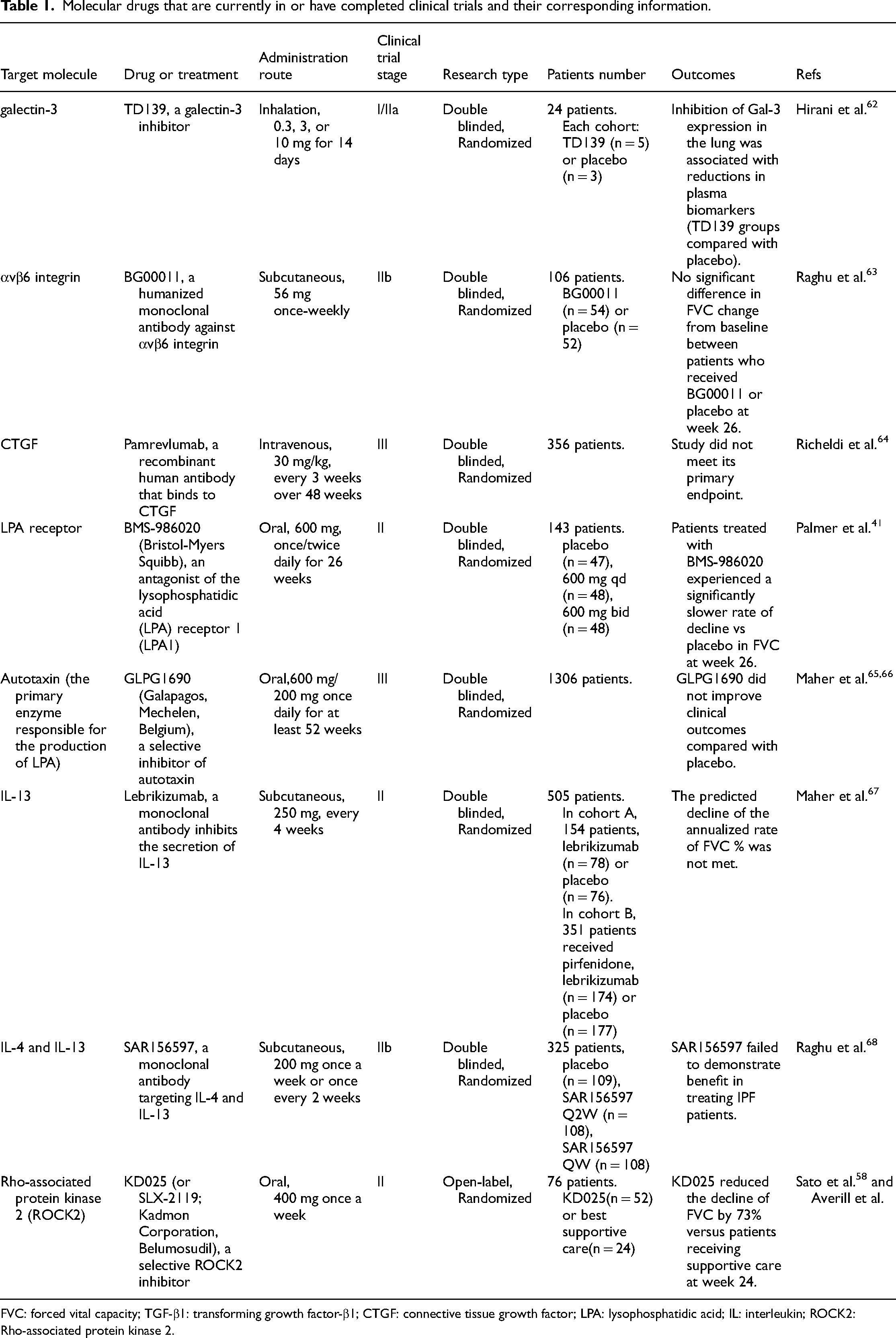
FVC: forced vital capacity; TGF-β1: transforming growth factor-β1; CTGF: connective tissue growth factor; LPA: lysophosphatidic acid; IL: interleukin; ROCK2: Rho-associated protein kinase 2.
TGF-β
TGF-β belongs to a family of evolutionarily conserved peptide factors essential to both embryonic and postnatal development.69,70 It is overexpressed in IPF and is one of the more intensively studied pro-fibrotic factors. Latent TGF requires activation by a few factors to be effective, such as integrins, 71 plasmin, 72 and thrombospondin 1. 73 Activated TGF-β regulates cellular gene expression through classical or non-classical pathways via interacting with the TGF-β receptor (TBR). 55
The TGF-β family includes TGF-β1, TGF-β2, and TGF-β3. 71 TGF-β1 is a pleiotropic cytokine that, in vitro, TGF-β1 inhibits the proliferation of alveolar epithelial cell (AEC) while promoting apoptosis. 74 In addition, TGF-β1 promotes epithelial-mesenchymal transition (EMT) 75 and induces activation of fibroblasts and subsequent differentiation to myofibroblasts. 3 TGF-β signaling can be blocked in four broad ways: firstly, to inhibit the production of TGF-β; secondly, to block its activation of latent TGF-β; thirdly, to neutralize the ligand or receptor; and fourthly, to interfere with the downstream pathways of TGF-β. 55
Inhibition of the TGF-β production
Serum amyloid P (SAP), also named Pentraxin 2, reduces the production of TGF-β1 and suppresses the conversion of peripheral blood monocytes to fibroblasts.29,76 In one research, injection of SAP into bleomycin-induced fibrosis mice attenuated structural damage to lung tissue, reduced collagen content production, and showed a significant decrease in fibroblast counts. 77 Based on a phase II clinical trial, patients receiving RhPTX-2/PRM-151 (recombinant human pentraxin 2) experienced a lower forced vital capacity (FVC) change of predicted value from baseline to week 28 than those receiving placebo. 29 However, the trial was terminated in 2023 because the drug was ineffective in the phase III clinical trial phase.
Blocking of the TGF-β activation
As a transmembrane protein, integrins are significantly involved in maintaining extracellular matrix adhesion and intercellular adhesion. The αvβ6 integrin, upregulated in BLM-induced pulmonary fibrosis, currently is one of the most studied potential therapeutic targets in IPF. It has been reported that αvβ6 integrin can bind TGF-β large latent complex (LLC), then lead to extracellular activation of TGF-β signaling. 78 Treatment of fibrotic mice with a functionally blocking anti-αvβ6 antibody markedly diminished pleural thickening and slowed the decline in pulmonary respiratory function by αvβ6 inhibition compared to fibrotic mice treated with a control antibody. 79 However, in a phase II clinical trial of an anti-integrin drug called BG00001, patients who received the drug did not have a significant change in FVC compared to placebo patients, and the trial was even terminated early because some patients experienced an acute exacerbation, suggesting extra pre-clinical studies are required to present such safety issues. 63
In addition to αvβ6, many integrins also regulate the activity of TGF-β and consequently promote the proliferation and growth of fibroblasts such as αvβ1 and αvβ3. These integrins are also expected to be targets for future research.
Neutralization of the TGF-β receptor
Galectin(Gal)-3 is a lectin that binds β-galactoside and is highly expressed in many fibrotic diseases. The β-galactoside-binding Gal-3 promotes fibrosis by regulating the expression of TGF-β receptors. Previous studies have reported that reduced TGF-β1-induced EMT, myofibroblast activation, and collagen deposition in Gal-3-deficient bleomycin-induced fibrosis mice can be observed. 80 Gal-3 expression was found to be downregulated in patients treated with TD139 (galectin-3 inhibitor) compared to controls in a phase I/IIa study. The results of this trial suggested that changes in IPF serum biomarkers (platelet-derived growth factor-BB, plasminogen activator inhibitor-1, CCL18, and YKL-40) in patients were associated with a downregulation of Gal-3. 62
Connective tissue growth factor (CTGF)
As a member of the CCN family with multiple biological effects, CTGF regulates cell proliferation, differentiation, adhesion, and various other biological processes. 81 This growth factor interacts with many components of the extracellular matrix and also binds to other growth factors to transduce signals, such as TGF-β and VEGF, 55 and can also exert a downstream effect via the mitogen-activated protein kinase (MAPK) pathway. 78 CTGF has a pro-proliferative and chemotactic effect on fibroblasts and promotes the accumulation of extracellular matrix, such as collagen type I and fibronectin. 82 Blockade of CTGF with FG-3019 reversed lung remodeling and restored lung function in mice with radiation-induced pulmonary fibrosis. 83 Pamrevlumab is a recombinant human antibody that binds to CTGF. In a phase II trial, patients treated with pamrevlumab experienced a noticeably lower decrease in FVC at week 48 than the placebo group. Also, a proportion of patients receiving pamrevlumab showed a lower rate of disease progression. 64 However, in the phase III trial, pamrevlumab was prematurely stopped in 2023 since the drug did not meet its primary endpoint.
VEGF, PDGF, and FGF
VEGF, PDGF, as well as FGF all belong to the ligands of tyrosine kinase receptors, and their abnormal expression in IPF has been reported previously.
There are seven members in the VEGF family: VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, placental growth factor, and snake venom vascular endothelial growth factors. 55 Among the VEGF family, VEGF-A is the most intensively researched member in relation to IPF. Regarding biological function, VEGF stimulates alveolar type II epithelial cell growth and angiogenesis, thereby helping epithelial and endothelial cells resist apoptosis and inhibit the development of IPF. 55 Still, Hamada's team found that mice with pulmonary fibrosis transfected with anti-VEGF genes had reduced lung collagen deposition, diminished pulmonary fibrosis, and showed anti-inflammatory and anti-angiogenic effects. 84 Consequently, the complex effects of VEGF in IPF are still controversial and require further exploration.
PDGF has the effect of inducing fibroblasts to secrete ECM components and growth factors, which also stimulates fibroblast multiplication and recruits them to the lung. 3 PDGF is phosphorylated upon binding to the receptor and activates downstream signaling pathways, such as Ras-MAPK and integrin-focal adhesion kinase (FAK) pathway. 55 Some studies have shown that PDGF expression is upregulated in pulmonary fibrosis mice model and that the progression of fibrosis in mice is retarded by using anti-PDGF antibodies. 78
Some of the FGF members were shown upregulated in the IPF. Secretion of FGF can be regulated by TGF-β, thereby inhibiting fibroblast recruitment and stimulating extracellular matrix production. After autophosphorylation of the FGF-binding receptor, various pathways can exert biological effects, including RAS-ERK, PI3K/AKT, PKC, and JAK-STAT signaling cascades. 78
In BLM-induced pulmonary fibrosis rats, the use of selective inhibitors of the family of VEGF-, PDGF- and FGF-receptor tyrosine kinases reduced lung fibrosis in BLM-treated rats relative to saline-treated rats, mainly by down-regulation of TGF-β1, pro-collagen-I expression. 85 Nintedanib, an intracellular multi-target tyrosine kinase inhibitor (TKI), targets vascular endothelial growth factor receptors (VEGFR) 1–3, fibroblast growth factor receptors (FGFR) 1-3, platelet-derived growth factor receptors (PDGFR) α and β, and non-receptor kinases like Src, all thought to be involved in IPF pathogenesis and was approved for IPF treatment in USA in October 2014.6,86,87 In a clinical study, the drug significantly improved the IPF patients’ lung function. 59
Lysophosphatidic acid (LPA)
LPA is a pro-inflammatory and pro-fibrotic molecule secreted by platelets during epithelial injury, activating six types of receptors (LPAR1-6) 3 and can mediate the migration of fibroblasts. 88 In IPF, LPA is expressed abnormally. We have identified that the LPA receptor 1 (LPA1) pathway was associated with IPF aberrant repair, facilitating fibroblast recruitment, increasing vascular permeability, inducing vascular leakage, and causing vascular endothelial cell dysfunction. 89 Compared to wild-type mice, LPA1-deficient mice showed significant protection from pulmonary fibrosis and fibroblast recruitment, as indicated by a remarkable change in fibroblast aggregation and significantly reduced collagen accumulation. 89 In a phase II trial, the change of FVC of patients who received BMS-986020, the antagonist of the LPA1, was substantially slower than that of the placebo group. 41
Autotaxin (ATX) is a nucleotide pyrophosphatase/phosphodiesterase (NPP) family member. It has been mentioned that ATX is mainly responsible for producing LPA in blood. 90 Some evidence suggested that concentrations of ATX in the lungs of patients with IPF and BALF from mice with pulmonary fibrosis increased. 58 GLPG1690 (Galapagos, Belgium), a selective inhibitor of ATX for oral administration, has been found to be associated with reduced concentrations of LPA in plasma in rats. 91 In a phase II study, patients in the GLPG1690 group showed an upward trend in the mean change in FVC at baseline, but patients who received placebo treatment, showed a significant decrease in FVC. 65 However, in a Phase III, randomized, double-blind, placebo-controlled, multicenter study designed to evaluate the efficacy and safety of two doses of GLPG1690, the trial was prematurely terminated due to a lack of benefit compared to placebo for the primary secondary outcome. 66
Interleukin (IL)-13
Epithelial damage leads to dysregulation of cytokine secretion from TH cells, and the related literature also reports the promotion of IPF by multiple cytokines, including IL-4, IL-8, IL-13, and so on.3,55 Notably, IL-13 has an important contribution to the development of the disease. IL-13 is able to activate fibroblasts, promote extracellular matrix synthesis, and induce myofibroblast transformation via TGF-β dependent or non-dependent pathways and apoptosis in epithelial cells.3,67 IL-13-deficient mice showed a reduction in fibrosis after BLM administration. 67 Regrettably, in a phase II trial of lebrikizumab, a monoclonal antibody binding to soluble IL-13, a predicted decline of FVC was not met in cohort lebrikizumab versus placebo. 67 This reminds researchers to make further enhancements.
Rho-associated coiled-coil forming protein kinase (ROCK) family
ROCK family belongs to serine/threonine kinases, which in human tissues can be divided into ROCK1 and ROCK2. 92 Some substances, such as LPA and thrombin, can activate the ROCK family, which is associated with the assembly and contraction of actin in cytoskeletal reorganization. Trauma repair in IPF can be traced back to the restructuring of the cytoskeleton, especially in the core cells involved in the disease, such as fibroblasts, epithelial cells, and endothelial cells. Following lung injury, the ROCK family becomes central to the downstream signaling pathway by various pro-fibrotic molecules in these cells. Treatment with selective RHO kinase inhibitors reduced lung hydroxyproline content and fibrosis in BLM-induced mice with pulmonary fibrosis. In an open-label phase II clinical trial, there was a significant change in FVC in IPF patients treated with KD025 (selective ROCK2 inhibitor) at week 24 when compared to the control group. 58
Janus kinases (JAK)/signal transducer and activator of transcription (STAT) pathway
The JAK/STAT pathway is commonly expressed in cells and is vital in cell differentiation, proliferation, and apoptosis. 8 In IPF, many pro-fibrotic factors, such as IL-13, TGF, and PDGF, act through the JAK/STAT pathway.8,93 JAKs, including JAK1, JAK2, and JAK3, are localized in the cell, and STATs, including STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6, are phosphorylated and enter the nucleus to regulate gene transcription. Related studies have shown that JAK1 and its active form are overexpressed in inflammatory and epithelial cells of lung tissue in the pulmonary fibrosis mouse model. 93 Following ablation of the JNK1 gene in bronchial and alveolar epithelial cells from BLM-induced fibrotic mice, fibrosis was suppressed compared to BLM-induced wild mice, mainly in the form of a reduction in hydroxyproline content and collagen, and a marked contrast in Masson's trichrome staining. 94 A multi-center, randomized, double-blinded, and placebo-controlled phase II study of Jaktinib dihydrochloride monohydrate (JAK1/2 inhibitors) is ongoing. The results of the trial (NCT04312594) are not further discussed in this work since the study on the primary outcomes is not completed yet.
Reactive oxygen species (ROS)
ROS, such as superoxide anion (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (OH•), could play immune defense roles in vivo, and can also act as second messengers to participate in the regulation of cellular signaling pathways and maintain the stability of the intracellular environment. All cells in the body can produce reactive oxygen species through the consumption and metabolism of oxygen. 95 In addition to the mitochondrial cellular respiratory chain that generates ROS, several enzymes such as xanthine oxidase, lipid peroxidase, uncoupled endothelial NO synthase, cytochrome P450 enzymes, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) are also involved in its synthesis. Under physiological conditions, proper ROS production contributes to normal cellular activity. However, in some disease states, overproduced ROS leads to a series of crosstalk imbalances. There is also dysregulation of ROS in IPF due to enhanced NOX expression and activation, as well as mitochondrial dysfunction and increased mtROS production. This imbalance leads to lung epithelial cell death, excessive collagen deposition, and persistent inflammation. 96 The NOX family consists of seven NOX homologs, NOX1-5, and the dual oxidases DUOX1 and 2. Currently, NOX4 is the most studied in relation to IPF and it is thought to be associated with fibrosis in many organs. On the one hand, NOX4 can act as a downstream signal for TGF-β to regulate the fibrotic process, and on the other hand, NOX-dependent redox signaling is also able to regulate the TGF-β/Smad signaling pathway in the form of feedback to indirectly promote the fibrotic process. 97 It has been found that NOX4-deficient mice exhibit significantly less lung epithelial cell death after bleomycin treatment compared to normal mice, suggesting that NOX4 plays a role in inducing at least epithelial cell death. 98 A randomized, double-blind, placebo-controlled phase II clinical trial of GKT137831 in patients with idiopathic pulmonary fibrosis is ongoing. This drug is an inhibitor of NOX isoforms.
Phosphodiesterase 4 (PDE4)
PDE4, an enzyme, belongs to the PDEs family, which has four isoforms, all of which are capable of mediating the hydrolysis of the second messenger, cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), to 5′-AMP and 5′-GMP, respectively, which in turn regulates downstream cellular activities. PDE4 mediates the production of pro-inflammatory and anti-inflammatory cytokines by regulating the degradation of cAMP. 99 PDE4 inhibitors have been reported to improve the sequelae of lung epithelial injury and significantly improve weight loss and collagen accumulation in the cellular matrix after administration of PDE4 inhibitors, either prophylactically or therapeutically, to mice modeling lung fibrosis caused by targeted injury to type II alveolar epithelium. 100 In another study, the effect of AA6216, a novel PDE4 inhibitor, was evaluated in a non-clinical IPF-related model and IPF patient samples. The results showed that therapeutic administration of AA6216 significantly reduced fibrosis scores, collagen-stained areas, and TGF-β1 production in BALF in a mouse model of bleomycin-induced pulmonary fibrosis. 101 They also found that AA6216 significantly inhibited the production of TNF-α by alveolar macrophages in IPF patients. 101 The antifibrotic effects of PDE4 inhibition are not fully understood. It is hypothesized that it may inhibit the transformation and proliferation of lung fibroblasts and the expression of extracellular matrix proteins. In a phase II, double-blind, placebo-controlled trial of BI 1015550, an oral preference inhibitor of the PDE4B subtype, in patients with idiopathic pulmonary fibrosis, a delay in change relative to baseline was found in patients using the drug at 12 weeks (FVC). 102
Most of the target molecules mentioned above were found to have more significant changes in patient metrics during the clinical trial phase, providing good evidence for the next step in the development of the trial. However, we can also see that both lebrikizumab, which targets the cytokine IL-13, and SAR156597, which dual targets IL-4 and IL-13, fail to exhibit significant effects at the primary efficacy endpoint (52 weeks) in clinical phase II, suggesting that blocking these two cytokines alone is not effective for improving the microenvironment of lung fibrosis in cells, so more subsequent trials are needed. In addition, most of the trials in Table 1 used monotherapy with placebo controls. However, many patients are now treated clinically with combination therapy with sound clinical outcomes. Therefore, the design of the drug combinations with FDA-approved drugs in further clinical trials may bring us more favorable treatment outcomes. As researchers explore, some evidence suggests that drugs for pulmonary fibrosis may improve lung cancer because of similarities in the pathogenesis of the two diseases, and based on this, it has become possible to use antifibrotic and antineoplastic drugs with each other or in combination to treat these highly fatal diseases. 103
Perspectives
The pathogenesis of IPF is complicated and involves a variety of cells, molecules, and signaling pathways (Figures 2 and 3). In this article, we mainly focused on the therapeutic targets that not only have entered clinical trials but also were publicly published with their clinical outcomes. Hence, a couple of pharmaceutical products are not discussed in our work, such as Human Umbilical Cord Mesenchymal Stem Cell Injection in phase I/IIa subject recruitment and ORIN1001 in phase I, since their final results have not been reported. Moreover, with the increasing understanding of the IPF mechanism, more and more potential therapeutic targets have been discovered, such as MMPs, 104 glycosaminoglycan, 105 non-coding RNA, 106 adenosine, 107 mechanosensitive MDM4, 108 micellar hyaluronidase, and spiperone. 109 So far, many researchers have focused on the downstream effectors of gene expression, such as cytokines and enzymes. The epigenetic modification also plays a vital role in IPF. Thus, we are looking forward to breakthroughs from this perspective in the future.
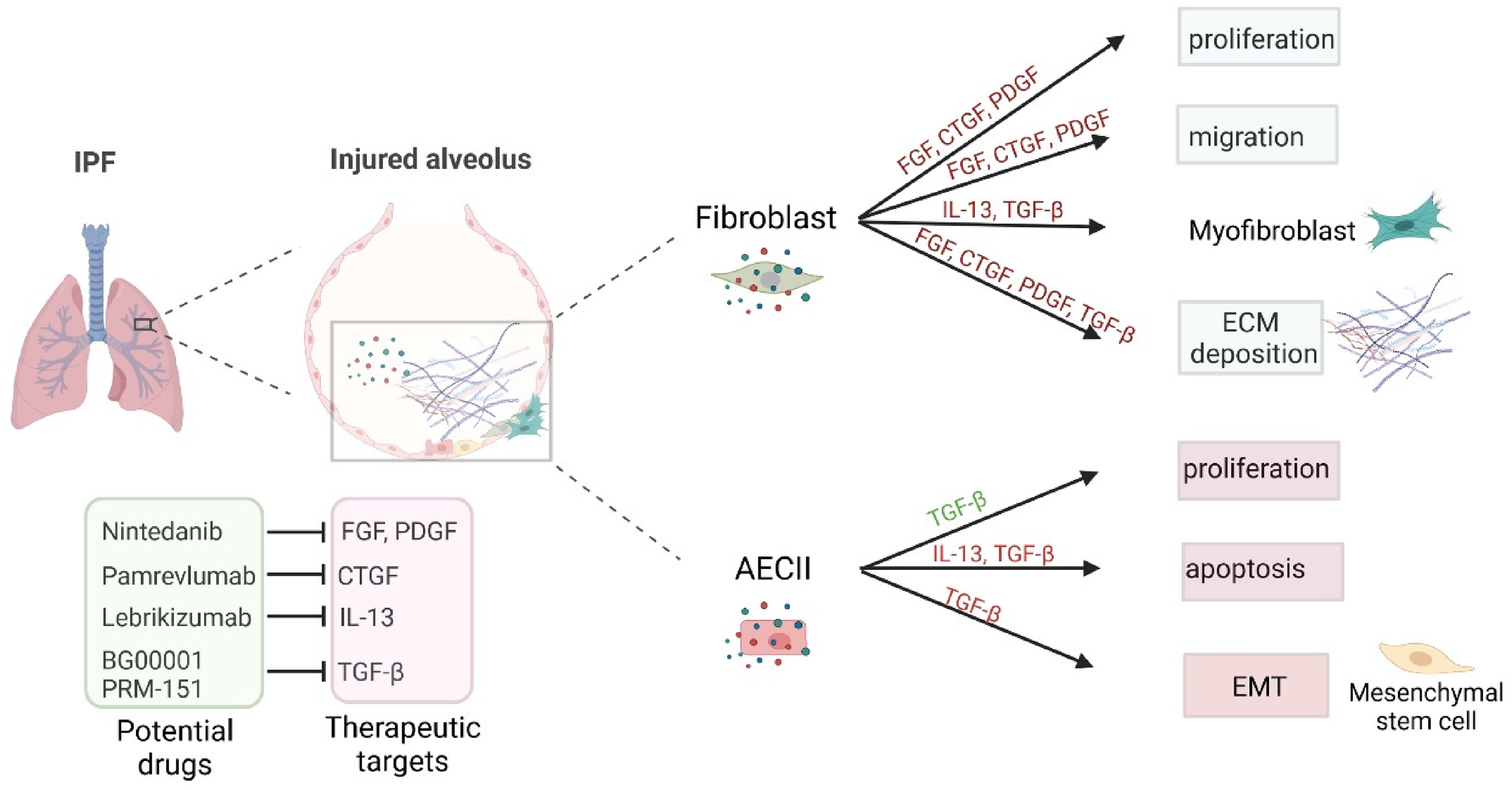
Molecular therapeutic products targeting IPF-related cytokines and growth factors.

Molecular therapeutics targeting the intracellular signaling cascades of IPF-related cytokines and growth factors.
Footnotes
Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (Grant Nos. 81970029 and 82171784), Shaanxi Province Natural Science Foundation (Project No. 2021JQ-024), and Xi'an Children's Hospital (2022A01 and 2020A03).
Author contributions
Yue Li: validation, visualization, and writing–original draft preparation; Congshan Jiang: validation, visualization, writing–original draft preparation, writing–reviewing and editing, and funding acquisition; Wenhua Zhu: validation, visualization, conceptualization, writing–reviewing and editing; Shemin Lu: conceptualization, funding acquisition, writing–reviewing and editing; Hongchuan Yu: conceptualization, writing–reviewing and editing, project administration, supervision; Liesu Meng: conceptualization, project administration, writing–reviewing and editing, and supervision.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China, Xi'an Children's Hospital, Natural Science Foundation of Shaanxi Province (grant number 81970029 and 82171784, 2022A01 and 2020A03, 2021JQ-024).
Author biographies
Yue Li, MSc candidate, Institute of Molecular Translational Medicine, Xi'an Jiaotong University. Yue Li's major is Immunology, and the research focuses on the pathogenesis of inflammatory diseases in the lungs.
Congshan Jiang, PhD, full researcher (research professorship) in the Pediatric Disease Research Institute of Xi'an Jiaotong University Affiliated Children's Hospital, has long been engaged in the molecular pathogenesis of chronic inflammatory diseases, as well as the construction and evaluation of animal models.
Wenhua Zhu, PhD, currently work as a professor in School of Basic Medical Sciences of Xi'an Jiaotong University. His research interest is the pathogenesis of chronic inflammatory diseases, especially focusing on innate immune regulation.
Shemin Lu, PhD and MD, Professor of Biochemistry and Molecular Biology from Xi'an Jiaotong University Health Science Center. Professor Lu has long been engaged in the molecular pathogenesis and susceptibility gene identification of chronic inflammatory diseases, as well as the construction and evaluation of animal models.
Hongchuan Yu, MD, Chief physician of pediatrics, from First Department of Respiratory Diseases, Xi'an Children's Hospital, Affiliated Children's Hospital of Xi'an Jiaotong University, specializing in respiratory diseases, skilled in fiberoptic bronchoscopy, and has been engaged in clinical and scientific research on pediatric respiratory diseases for 27 years.
Liesu Meng, PhD and MD, Professor of Biochemistry and Molecular Biology from Xi'an Jiaotong University Health Science Center. Professor Meng has long been engaged in the molecular pathogenesis and transformation of chronic inflammatory diseases, as well as the molecular and translational medicine.