
Abstract
Treacher Collins syndrome (TCS) is a rare congenital craniofacial disorder, typically inherited as an autosomal dominant condition. Here, we report on a family in which germline mosaicism for TCS was likely present. The proband was diagnosed with TCS based on the typical clinical features and a pathogenic variant TCOF1 (c.4369_4373delAAGAA, p.K1457Efs*12). The mutation was not detected in his parents’ peripheral blood DNA samples, suggesting a de novo mutation had occurred in the proband. However, a year later, the proband's mother became pregnant, and the amniotic fluid puncture revealed that the fetus carried the same mutation as the proband. Prenatal ultrasound also indicated a maxillofacial dysplasia with unilateral microtia. The mother then disclosed a previous birth history in which a baby had died of respiratory distress shortly after birth, displaying a TCS-like phenotype. Around the same time, the proband's father was diagnosed with mild bilateral conductive hearing loss. Based on array data, we concluded that the father may have had germline mosaicism for TCOF1 mutation. Our findings highlight the importance of considering germline mosaicism in sporadic de novo TCOF1 mutations when providing genetic consulting, and prenatal diagnosis is important when the proband's parents become pregnant again.
Introduction
Treacher Collins syndrome (TCS, OMIM 154500), also known as Franceschetti syndrome or mandibulofacial dysostosis (MFD1), is a rare disorder of craniofacial development with an estimated birth prevalence of 1 in 50,000 live births. 1 Diagnosis of TCS is based on clinical, familial, and genetic factors. The most common clinical features include antimongoloid slant of the eyes, coloboma combined with various degree of absence of eyelashes of the lower eyelids, complex hypoplasia of the mandible and zygomatic arch, microtia, macrostomia, and micrognathia. Additional symptoms such as cleft lip and palate, conductive hearing loss due to ossicle deformity, dyspnea, feeding difficulties, and developmental delays may also occur. TCS is typically inherited as an autosomal dominant condition and is thought to exhibit nonpenetrance, with 60% of cases occurring as a result of de novo mutations, while the remaining 40% involve a family history.2,3
It is widely acknowledged that TCS may show an intra- and inter-familial phenotypic variability, with some cases being severely affected and experiencing respiratory distress after birth, while others may be mildly affected and go undiagnosed. Somatic variants can spontaneously occur in any cells of the body and are usually limited to the descendants of the mutated original cell and will not be passed on to the next generations. In contrast, germline variants may occur in the germ cells and can therefore be passed on to the next generations. When couples who have given birth to a child with a genetic disorder or have a family history of such disorders have a desire for further reproduction, they often seek genetic counseling. In more than 60% of patients with TCS, the variant occurs “de novo,” and parents of the proband are asymptomatic, with the variant not being detectable in their peripheral blood. Nevertheless, several reports in the literature have described affected siblings born to unaffected parents, which may be due to gonadal mosaicism.4,5 Here, we report the first case of three successive births resulting in TCS in nearly healthy parents in China. Our aim with this study is to emphasize the possibility of mosaic variant when explaining a de novo variant in families affected by TCS.
Case report
Personal history
The proband (III:2, Figure 1) of this family was born with typical TCS phenotype, including antimongoloid slant of the eyes, coloboma, and absence of eyelashes of the lower eyelids, complex hypoplasia of the mandible and zygomatic arch and bilateral microtia atresia. At 6 years of age, the proband was referred to our facility for hearing improvement. The pure tone audiometry test showed a bilateral severe conductive hearing loss (Figure 2(a)). The temporal computerized tomography (CT) indicates bilateral malformation of ossicular chain, with the right auditory ossicles absent, and the left malleus and stapes not clearly displayed, and the craniofacial radiographs reconstruction suggest abnormal development of maxilla, zygomatic bone, and mandible (Figure 2(b)). The research was carried out in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of Peking Union Medical College Hospital (no. JS-796). Written informed consent for the publication of this study was provided at the clinic on August 6, 2022 by the patient's parents.

Pedigree of the TCS. Boxes indicate male subjects; circles indicate female subjects. Black boxes/circles indicate the presence of the TCS. Gray circles indicate highly possible TCS. The oblique line indicates that the respective subject has deceased. The small black box in patient II:3 indicates the presence of germline and somatic mosaicism. Arrow points at proband. TCS: Treacher Collins syndrome.
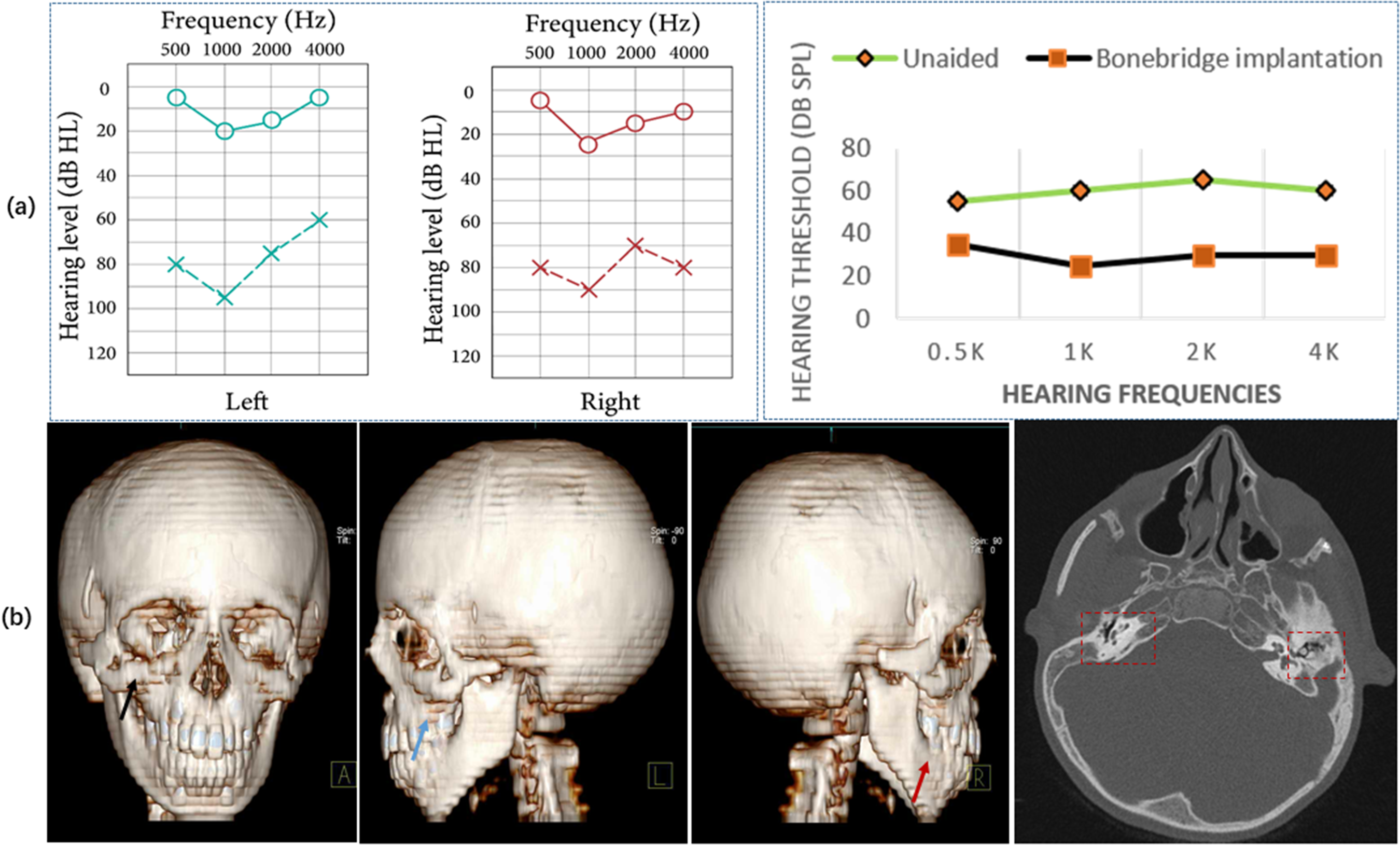
(a) The pure tone audiometry test showed bilateral severe conductive hearing loss. And the hearing threshold with and without Bonebridge. (b) The craniofacial radiographs reconstruction suggests abnormal development of maxilla (dark line), zygomatic bone (blue line), and mandible (red line), in maxillofacial region. The temporal computerized tomography indicates that the right auditory ossicles were absent, and the left malleus and stapes were not clearly displayed. (c) The TCOF1 deletion (c.4369_4373delAAGAA, p.K1457Efs*12) in DNA extracted from peripheral blood of the proband, which was undetected in the parents’ peripheral blood. The amniotic fluid puncture revealed that the fetus carried the same variant as the proband.
Genetic testing
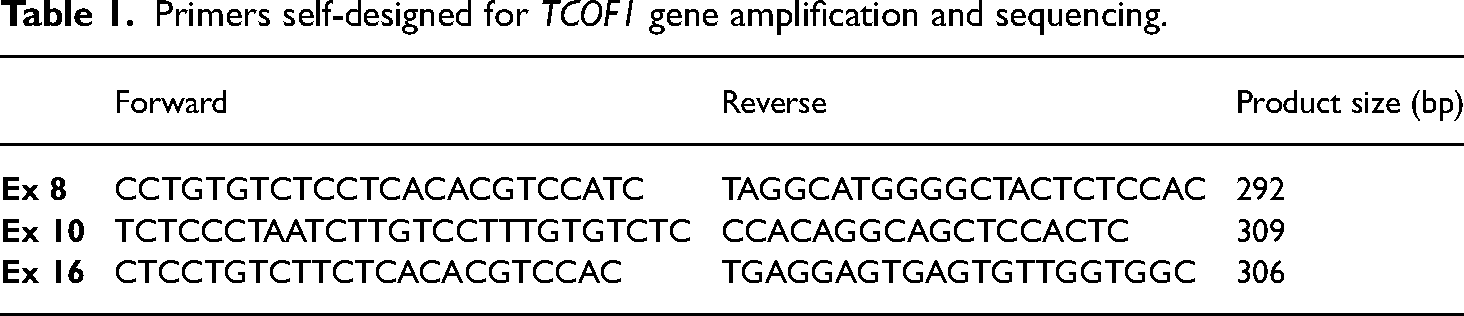
The genomic DNA extraction from peripheral blood samples was obtained with TIANamp Blood DNA Kit (Tiangen) according to the manufacturer's protocol. The 27 coding exons (including exons 6A, 16A and 19) and intron/exon boundaries of TCOF1 gene were amplified in 28 fragments. Mostly amplification primers were previously reported,6,7 and primers of exons 8, 10, and 16 were self-designed (Table 1). The coding regions and intron/exon boundaries of POLR1D and POLR1C genes were amplified with primers previously reported.8,9 The PCR products were purified with Beckman Coulter Agencourt AMPure XP kit (Beckman Coulter), and were subsequently subjected to Sanger sequencing with ABI3730xl DNA Sequencer. The resulting sequences were analyzed using the Sequencing Analysis 5.2 software (Applied Biosystems). The reference sequence used for comparing of TCOF1, POLR1D, and POLRIC genes were NM_001135243.1, NM_015972.3, and NM_203290.2, respectively. Nomenclature for the variants is based on Human Genome Variation Society (HGVS) nomenclature guidelines (http://www.hgvs.org/mutnomen).
Primers self-designed for TCOF1 gene amplification and sequencing.
Results of genetic testing
DNA analysis in the proband showed the presence of a TCOF1 variant (c.4369_4373delAAGAA, p.K1457Efs*12) (Figure 2(c)), which has been known as the true hot spot. 10 The parents did not exhibit any discernible TCS phenotype and reported no familial history of the condition. Additionally, the variant was not detected in their peripheral blood-derived DNA samples. Based on these findings, we inferred that the variant in the proband was likely to be a de novo variant.
Family history
Subsequently, approximately 1 year later, the mother of the proband became pregnant, and the amniotic fluid puncture revealed that the fetus carried the same variant as the proband, indicating the presence of germline mosaicism in one of the proband's parents. In addition, the prenatal ultrasound also indicated a maxillofacial dysplasia with bilateral microtia. At this point, the mother disclosed a previous birth history wherein the baby had died due to respiratory distress after birth and exhibited a TCS-like phenotype. However, no genetic diagnosis was made at that time, and the symptoms were only recalled from the mother's memory. A comprehensive clinical examination was subsequently conducted on both parents, which revealed that the father had a mild bilateral conductive hearing loss that had been ignored due to its mild nature and negligible impact on daily life. Furthermore, the temporal CT scan showed bilateral ossicular chain malformation, which might be one of the mild phenotypes associated with TCS. Based on these findings, we concluded that the father might have a high rate of gonadal mosaicism.
Hearing intervention and the outcome
The proband was diagnosed with bilateral severe conductive hearing loss according to the pure tone auditory test and a bone conductive hearing device is a proper choice. After many considerations, the patient chose bonebridge implantation surgery. The CT showed enough space for bonebridge implantation in the right mastoid. The bonebridge implantation surgery was conducted at our hospital, and a 6-year follow up has been completed to date (current age 12 years). The hearing test results suggest that the bonebridge implantation has resulted in an optimal hearing outcome for this patient (Figure 1(a)).
Discussion
TCS is classified into four clinical subtypes based on the variants observed. TCS1 (OMIM 154500) is caused by variants in the treacle ribosome biogenesis factor 1 (TCOF1) gene, TCS2 (OMIM 613717) is caused by variants in the RNA polymerase I (pol I) subunit D (POLR1D) gene, TCS3 (OMIM 248390) is caused by variants in the RNA pol I subunit C (POLR1C) gene, and recently identified pathogenic variants of RNA pol I subunit B (POLR1B) gene are classified as TCS4 (OMIM 618939). 10 This syndrome is genetically heterogeneous, with an autosomal dominant inheritance pattern observed in TCOF1, POLR1D, and POLR1B, while a recessive pattern is observed in POLR1D and POLR1C.
The TCOF1 gene, which contains 27 exons, encodes the Treacle protein. To date, over 200 pathogenic variants have been identified in TCOF1, including deletions, insertions, splicing, and substitutions. The variant hotspots of TCOF1 are located in exons 10, 15, 16, 23, and 24, and most pathogenic variants in these regions result in a frame-shift, leading to a termination codon and haploinsufficiency.11,12 There is a recognized nuclear import signal located at the C-terminus and a nuclear export signal located at the N-terminus of Treacle, which plays a vital role in regulating its dynamic localization and is involved in the transport of ribosomal subunits between the nucleolus and cytoplasm. Treacle regulates ribosomal DNA gene transcription by binding and interacting with the upstream binding factor (UBF), Pol I, and Nopp140 at the sites of rDNA promoter. Decrease expression of treacle due to TCOF1 variant can result in the rapid displacement of UBF from rDNA, which can inhibit the rRNA synthesis. Conversely, an increase in Treacle levels may interfere with cisplatin-induced apoptosis. Therefore, both reduced expression and abnormal distribution of treacle can lead to apoptosis of cranial neural crest cells during embryogenesis by influencing crucial apoptotic regulators.13,14 POLR1D and POLR1C are subunits of shared RNA polymerases I and III, which play a crucial role in ribosomal RNA transcription. Insufficient amounts of Pol I or Pol III due to variants in POLR1D or POLR1C can result in a reduced number of mature ribosomes in cranial neural crest cells during embryonic development. This can affect the normal apoptosis of the first and second branchial arches, resulting in craniofacial abnormalities. On the other hand, POLR1B is a unique subunit of RNA polymerase I, and pathogenic variants in this gene can cause craniofacial defects by inducing p53-dependent apoptosis.8,11
Approximately 60% of TCS cases arise from a de novo variant. While this percentage is typical for an autosomal dominant inherited disease, mosaicism may occur in some of these cases. TCS is also known to exhibit variability, which is often described as nonpenetrance. However, some of this variation may also be attributed to mosaicism. 15 Variants occurring during later stages of embryonic development in differentiated cells lead to a phenotype limited to the specific affected tissue, known as somatic mosaicism. Somatic variants are typically observed and reported in sporadic tumors. 16 If a variant occurs before the separation into germinal cells, it may be present in both somatic and germ cells. Individuals with somatic and germline mosaicism may exhibit varying degrees of symptoms and have a risk of passing the variant on to their offspring. Mosaicism can be classified as somatic, affecting only somatic cells and not the germline, or gonosomal, affecting both somatic cells and the germline. In the context of genetic counseling, it is recognized that germline mosaicism can occur for autosomal dominant disorders, and the risk of recurrence is typically estimated to be between 1% and 6% for apparent de novo variants.17,18 In this study, we report on a family in which combined somatic and germline mosaicism for TCOF1 was suspected. Three siblings in the family exhibited the TCS phenotype, and two of them were found to carry the same TCOF1 variant. The third sibling was not tested for the gene but is likely to carry the same variant. Interestingly, the variant was not detected in the peripheral blood of the parents, suggesting that it was present as somatic and germline mosaicism in one or both of the parents.
While TCS is typically considered an autosomal dominant disease, there is growing evidence to suggest that inheritance patterns may be more heterogeneous. The case presented in this study supports our hypothesis that mosaicism, specifically, maybe a contributing factor to the wide phenotypic variability observed in TCS. Furthermore, it is possible that some instances of nonpenetrance in TCS may be attributed to mosaicism. To our knowledge, there is only one previous report of somatic mosaicism of TCOF1 in TCS. 4 In that study, a TCOF1 1408delAG heterozygous variant was identified in a patient with TCS, and the same variant was found in the clinically unaffected mother's leukocytes, hair root bulbs, buccal mucosa, urine, and stool, but not in her skin fibroblasts. The mother had a clinically unaffected child, and the maternal grandparents did not have the variant. The authors concluded that the mother was mosaic due to cell type-specific selection. While affected siblings born to parents without phenotype have been reported, an autosomal recessive form has also been proposed as a possible explanation, as seen in other mandibulofacial dysostosis diseases. 19 In our family, as the same variant was identified in more than one sib, we speculated that one of the parents is likely to have germline mosaicism. Given the clinical features, conductive hearing loss due to ossicular malformation, present in the father of the proband, we suspect that the father may have somatic mosaicism. However, characterizing mosaicism can be challenging due to several limitations. The inaccessibility to genital tissues precluded the assessment of the extent of gonadal mosaicism. Additionally, it is difficult to determine the specific tissue in which the variant occurs unless accompanied by an associated phenotype. Although it may be easier to identify somatic mosaicism when the variant is detected in a specific tissue, it is difficult to confirm its existence without evaluating every tissue type and cell. Finally, the temporal and spatial occurrence of the variant during development makes it challenging to evaluate mosaicism. 20
Conclusion
In summary, this is the first reported case of three successive births with TCS to nearly unaffected parents. We suspect that the father may have a high rate of gonadal mosaicism based on the observed pattern of inheritance. Additionally, this case report highlights the importance of considering the possibility of mosaicism in genetic counseling for apparently sporadic de novo TCOF1 variants, and prenatal diagnosis is of significance when the proband's parents become pregnant again. This is essential for accurate risk assessment of offspring and to inform appropriate management strategies.
Footnotes
Acknowledgements
We appreciate all the participants for their involvement in the study.
Authors’ contributions
XC conceived and designed the experiments, and acted as the attending physician of the proband. XF and TY collected the data and wrote the article. XL and YC assisted in conducting and analyzed the CT data. XC and TY revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript and collected data.
Data availability
All data generated during this study are included in this published article and its supplementary information files.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical statement
The research was carried out in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of Peking Union Medical College Hospital (no. JS-796). Written informed consent for the publication of this study was provided by the patient's parents.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National High Level Hospital Clinical Research (grant number No.2022-PUMCH-D-002 and 2022-PUMCH-C-029).