
Abstract
Caylusea hexagyna and Ochradenus baccatus are two species in the Resedaceae family. In this study, we analysed the complete plastid genomes of these two species using high-throughput sequencing technology and compared their genomic data. The length of the plastid genome of C. hexagyna was 154,390 bp while that of O. baccatus was 153,380 bp. The lengths of the inverted repeats (IR) regions were 26,526 bp and 26,558 bp, those of the large single copy (LSC) regions were 83,870 bp and 83,023 bp; and those of the small single copy (SSC) regions were 17,468 bp and 17,241 bp in C. hexagyna and O. baccatus, respectively. Both genomes consisted of 113 genes: 79 protein-coding genes, 30 tRNA genes and 4 rRNA genes. Repeat analysis showed that the plastid genome included all types of repeats, with more frequent occurrences of palindromic sequences. Comparative studies of SSR markers showed that there were 256 markers in C. hexagyna and 255 in O. baccatus; the majority of the SSRs in these plastid genomes were mononucleotide repeats (A/T). All the clusters in the phylogenetic tree had high support. This study reported the first complete plastid genomes of the genera Caylusea and Ochradenus and the first for the Resedaceae family.
Introduction
Resedaceae S.F. Gray is the smallest family in the Brassicales comprising 107 species in 12 genera, 1 and is distributed in arid to subtropical regions of Europe, Asia, the Middle East, North America, Mesoamerica, the Caribbean, India and Africa.2,3 Resedaceae was in the order Rhoedales, 4 Later, Rhoedales was renamed to Resedales. 5 Resedaceae was then moved to the order Capparidales. 6 This order (Capparidales) was renamed taxonomically into Capparales.7–10 Finally, the order Capparales was renamed as Brassicales.11–14 Serval taxonomic studies have moved some genera from other families into the Resedaceae family such as Borthwickia (formerly Borthwickiaceae), Neothorelia, Stixis and Tirania (formerly Stixidaceae) and Forchhammeria (formerly in Capparaceae) which has caused the Resedaceae family to expand.13,14
Caylusea hexagyna (Forsk.) M. L. is a green annual or perennial herb that is distributed in North and East Africa, the Middle East and Iran. 15 Ochradenus baccatus Delile is a perennial shrub that is distributed in Ethiopia, Egypt, Libya, the Middle East, Iran and parts of Pakistan.16–18 Ochradenus baccatus has antioxidant and anti-inflammatory activities, with uses in the treatment of diarrhea and colonic ulceration. 19
Chloroplast genomes are similar across most angiosperms; in terms of their coding regions, sizes, tRNA genes, and other related genes, even though the nature of chloroplasts may vary, as reported by different authors.20–22 Complete chloroplast genomes have been used as a vital tool for re-examining the phylogenetic relationships of complex taxa; these genomes have recently been used to resolve some unanswered questions in plant taxonomy. 23
This study provides the first insights into the systematic position of two species (C. hexagyna and O. baccatus) and provides valuable resources for further phylogenetic and evolutionary relationship studies of the species in the Resedaceae family to resolve the taxonomic problems of species such as: Borthwickia, Neothorelia, Stixis, Tirania and Forchhammeria.
Materials and methods
Plant material and DNA extraction
Fresh young leaf materials for C. hexagyena were collected from the Al-Shafa region (21.1561°N 40.3672°E) and those for O. baccatus were collected from the Al-Taif region (21.2999°N 40.3768°E), Saudi Arabia. Total genomic DNA was extracted from the samples using a Qiagen genomic DNA extraction kit (Qiagen, Inc., Germany) according to the manufacturer's protocols.
Library construction, sequencing and assembly
A total amount of 1.0 μg DNA was used as input material for the DNA sample preparations. The NEBNext DNA Library Prep Kit was used to generate sequence libraries according to the manufacturer's recommendation; indices were also added to each sample. Genomic DNA was randomly fragmented by shearing to a size of 350 bases in length. The ends of randomly fragmented DNA were repaired and A-tailed, adapters were ligated with NEBNext for Illumina sequencing, then the PCR improved by P5 and indexed P7 oligo sequences. The AMPure XP system was used to purify the PCR products; subsequent findings were analyzed by the Agilent 2100 Bioanalyzer for size distribution and later quantified using real-time PCR. 22 The total genomic DNA was sequenced using an Illumina Hiseq 2500. The raw reads data were subjected to PRINSEQlite to obtain clean data (5Gb). The generated reads were assembled using NOVOPlasty 24 with kmer (K-mer = 33) to assemble the complete chloroplast genome from the whole genome sequence. The ndhF intergenic spacer from Cleome chrysantha (KF923088) was used as the seed, and the plastome sequence of Stapelia gigantea (MG963259) was used as the reference for the assembly of the C. hexagena cp genome. For O. baccatus, the accD gene from Brassica juncea (JF801163) was used as the seed, and the plastome sequence of Brassica juncea (NC_028272) was used as the reference. Finally, for each species, one contig containing the complete chloroplast genome sequence was generated.
Gene annotation
In C. hexagyna and O. baccatus genes were annotated using DOGMA (Dual Organellar GenoMe Annotator, University of Texas at Austin, Austin, TX, USA). 25 The positions of start and stop codon were adjusted manually. tRNA genes were identified by the trnAscan-SE server (http://lowelab.ucsc.edu/tRNAscan-SE/). 26 The circular chloroplast genome maps were drawn using OGDRAW (Organellar Genome DRAW). 27 The sequences of the chloroplast genome were deposited in the GenBank database: C. hexagyna (MT948187) and O. baccatus (MT948189).
Sequence analysis
The relative synonymous codon usage (RSCU) values, base composition and codon usage were analysed using MEGA 6.0 software. Potential RNA editing sites present in the protein-coding genes were predicted by the PREP suite 28 with the cut-off value set to 0.8.
Repeat analysis of the chloroplast genome
The online software MIcroSAtellite (MISA) 29 was used to identify simple sequence repeats (SSRs) in the chloroplast genome with the following parameters: eight, five, four and three repeat units for mononucleotide, dinucleotide, trinucleotide and tetra-, penta-, and hexa-nucleotides SSR motifs, respectively. REPuter software (https://bibiserv.cebitec.uni-bielefeld.de/reputer) 28 was used with default settings to detect the size and location of the long repeats in the two Cleomaceae species.
Genome comparison
Comparisons were made against the border region between inverted repeat (IR), large single copy (LSC) and small single copy (SSC) among the two species of Resedaceae chloroplast genomes.
Characterization of substitution rate
The nonsynonymous (dN) and synonymous (dS) substitution rates were calculated using DNAsp v5.10.01. 30 The chloroplast genome of C. hexagyna was compared with the chloroplast genome of O. baccatus to identify the genes that are under selective pressure. Geneious software v. 8.1.3 (Biomatters, Ltd, Auckland, New Zealand) was used to align the individual protein-coding genes separately, and the aligned sequences were then translated into protein sequences.
Phylogenetic analysis
The analysis was conducted based on the complete chloroplast genome sequences of two Resedaceae species (C. hexagyna and O. baccatus), three Capparaceae species, four Cleomaceae species, eight Brassicaceae species and two Malvaceae species, which were used as outgroups. All of the sequences were aligned using MAFFT 31 with default settings. The phylogenetic trees were reconstructed based on the maximum parsimony (MP) method using PAUP version 4.0b10 32 and a heuristic search strategy of 1000 random sequence addition replicates with tree bisection– reconnection (TBR) branch swapping. A maximum of 100 trees per replicate were saved, with MulTrees turned on and gaps treated as missing data. Statistical support was assessed for clades with nonparametric bootstrap analysis using 1000 bootstrap replicates. A 50% majority-rule consensus tree was calculated from all the most parsimonious trees. Bayesian inference (BI) analysis were performed in MrBayes version 3.2.6, 33 and the GTR models were selected using jModelTest version 3.7. 34
Results
Characteristics of C. hexagyna and O. baccatus chloroplast genomes
The complete chloroplast genome sequence has a circular and quadripartite structure. The total length of the C. hexagyna chloroplast genome is 154,390 bp, and that of O. baccatus is 153,380 bp. The chloroplast genome has four distinct regions: a large single copy (LSC), which a length of 83,870 bp in C. hexagyna and 83,023 bp in O. baccatus, a small single copy (SSC) region with a length of 17,468 bp in C. hexagyna and 17,241 bp in O. baccatus; and a pair of inverted repeats (IRa and IRb) that separate the SSC and LSC with lengths of 26,526 bp in C. hexagyna and 26,558 bp in O. baccatus (Figure 1). The region coding for genes C. hexagyna is 76, 620 bp in length, which constitutes 59.62% of the chloroplast genome, in O. baccatus, this region is 77,268 bp in length, which constitutes 50.37% of the chloroplast genome. The remainder of the chloroplast genome is the noncoding region, which includes introns and intergenic spacers with lengths of 68,293 bp (44.23%) in C. hexagyna and is 67,177 bp (43.79%) in O. baccatus. The GC percentage in the inverted repeat regions is higher than that in the large single copy and small single copy regions. The IRa and IRb regions have a GC content of 42.2% in both species, while the LSC and SSC regions have GC contents of 34.25% and 29.8%, respectively, in C. hexagyna and 34.16% and 29.49%, respectively, in O. baccatus (Table 1).

Gene map of the C. hexagyna and O. baccatus chloroplast genomes. Genes shown outside the circles are transcribed in the counter-clockwise direction, and those inside the circles are transcribed in the clockwise direction. The coloured bars indicate functional genes. In the inner circle, the dark grey area indicates the GC content, while the light grey colour indicates the AT contents. LSC indicates the large single copy region; SSC, indicates the small single copy region, and IR indicates inverted repeats.
Base composition in the C. hexagyna and O. baccatus chloroplast genomes.

The complete chloroplast genomes of C. hexagyna and O. baccatus contained a total of 134 genes, 113 were unique, and 19 genes were duplicated in the pair of IR regions. There were 79 protein-coding genes, 30 tRNAs and four rRNAs in the chloroplast genomes (Figure 1 and Table 2). Eight protein-coding genes were located in the IR region, along with seven tRNAs and four rRNAs. Sixty protein-coding genes and 22 tRNA genes were located in the LSC region, while the SSC region contained 12 protein-coding genes and one tRNA. Similar to most flowering plants (angiosperms), all the protein-coding genes in the chloroplast genome start with methionine (ATG codon), whereas some genes start with other codons, such as ATC, GTG and ACG.35–37
Gene contents in the chloroplast genomes of C. hexagyna and O. baccatus.

Gene with one intron, ++ Gene with two introns and a Gene with multiple copies.
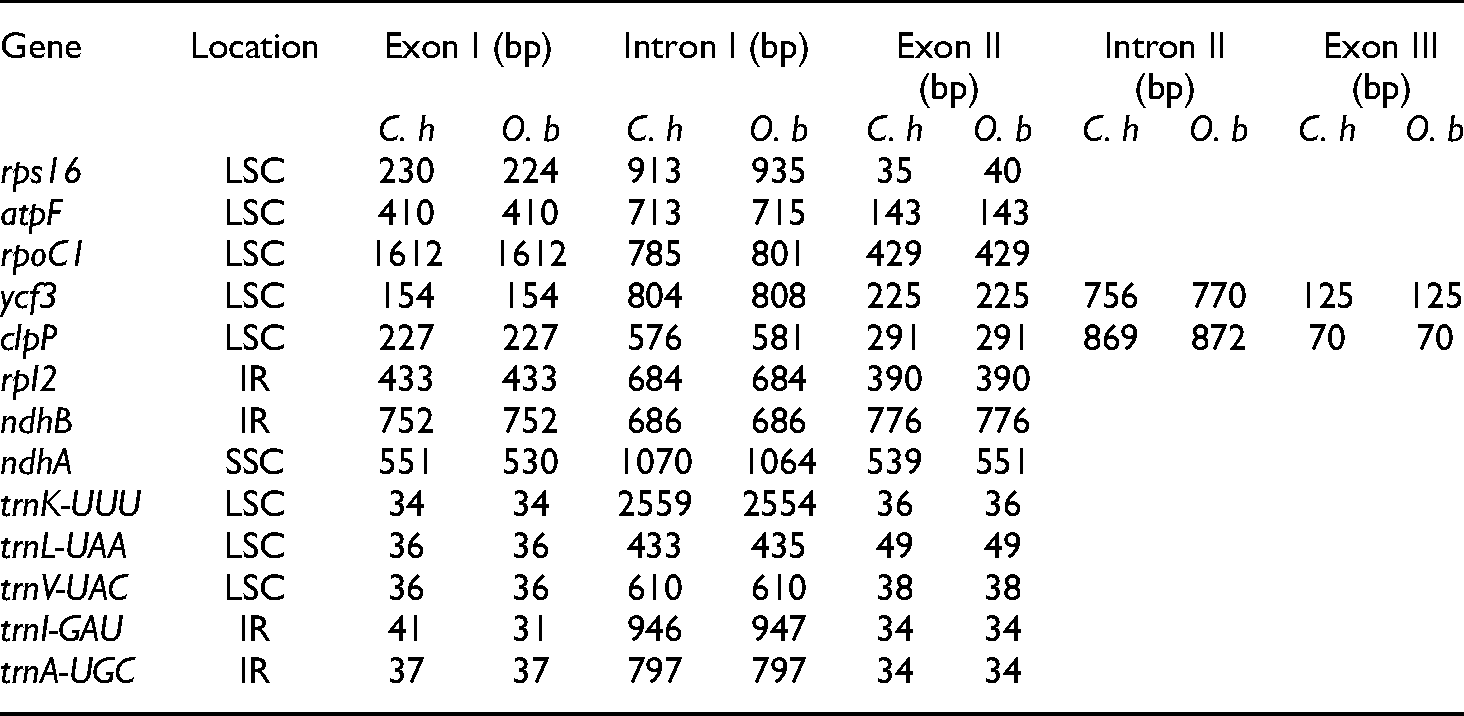
Similar to those in all angiosperms, the chloroplast genomes in both species contained introns in some of the protein-coding and tRNA genes.35,36 Among the 113 unique genes, 13 contained introns, eight of those were protein-coding genes and five were tRNAs (Table 3). Eight genes were found in the LSC region, with four genes in the IR region and only one gene in the SSC region. Eleven genes had only one intron, and two genes had two introns, ycf3 and clpP. The trnK-UUU gene had the longest intron (2559 bp in C. hexagyna and 2554 bp in O. baccatus) (Table 3).
Length of introns and exons in the C. hexagyna and O. baccatus chloroplast genomes.
Codon usage
The nucleotide sequences of protein-coding genes and tRNA genes were used to compute the frequency of the codon usage in chloroplast genomes; these sequence lengths were 79,613 bp in C. hexagyna and 80,236 bp in O. baccatus. The relative synonymous codon usage of the genes in these genomes is presented in S1 and S2 Tables. The results showed that the genes in the chloroplast genome were encoded by 26,439 codons in C. hexagyna and by 26,646 codons in O. baccatus. Codons coding for leucine were the highest in both genomes, with 2719 (10.28%) in C. hexagyna and 3697 (13.87%) in O. baccatus, whereas those coding for cysteine were the least abundant, with 325 (1.22%) in C. hexagyna and 477 (1.79%) in O. baccatus (Figure 2). The results (S1, S2 Table) showed that all amino acids have codon usage bias, except for tryptophan and methionine, which have RSCU values of 1.

Amino acid frequencies in the protein-coding sequences of C. hexagyna and O. baccatus chloroplast genomes protein-coding sequences.
RNA editing sites
The program PREP suite was used to predict the RNA editing sites in both chloroplast genomes. The first nucleotide of the first codon was used in all analyses. The results showed that most of the conversions in the codon positions were from the amino acid serine to that of leucine (S3, S4 Table). In total, the program revealed 41 editing sites distributed within 15 protein-coding genes in the genome of C. hexagyna and 39 editing sites distributed within 14 protein-coding genes in the genome of O. baccatus. In both genomes: ndhD had the highest number of editing sites (nine sites), ndhB had seven editing sites, and ndhF had five editing sites, the remaining genes ranged from four to one editing site: accD, atpF, clpP, matK, ndhA, ndhG, psaI, rpoB, rpoC2, rps2 and rps14 in both genomes and rps16 in C. hexagyna (S3, S4 Table). Conversions of proline (nonpolar) to serine (polar) were observed in the RNA editing site. The remaining genes (atpA, atpB, atpI, ccsA, petB, petD, petG, petL, psaB, psbB, psbE, psbF, psbL, rpl2, rpl20, rpl23, rpoA, rpoC1, rps8 and ycf3, in either genome and rps16 in O. baccatus) do not have predicted RNA editing sites in the first nucleotide of the first codon.
Repeat analysis
Long repeats
The program REPuter was used to identify long repeat sequences present in the C. hexagyna and O. baccatus chloroplast genomes using default settings; from the results, it was discovered that all four types of repeats (palindromic, forward, reverse and complement) were present in both chloroplast genomes (S5, S6 Table). The analysis of C. hexagyna and O. baccatus showed 20 and 22 palindromic repeats, 11 and 10 forward repeats, 14 and 12 reverse repeats and 4 and 5 complement repeats, respectively (S5, S6 Table and Figure 3). In total, there were 49 repeats in both chloroplast genomes. The majority of the repeat sizes in C. hexagyna were between 10 and 19 bp (51.02%), followed by those between 20 and 29 bp (38.77%) and 30 and 39 bp (10.2%); in O. baccatus, the majority of the repeat sizes were between 20 and 29 bp (93.87%), followed by those between 30 and 39 bp (6.12%). The intergenic spacer region in C. hexagyna and O. baccatus harboured 65.3% and 83.67% of the repeats, respectively. In C. hexagyna, the tRNA region contained nine repeats (18.36%), while 12 repeats (24.48%) were located in the protein-coding genes; in O. baccatus, the tRNA region contained five repeats (10.2%), and the protein-coding genes contained six repeats (12.24%) (S5, S6 Table).
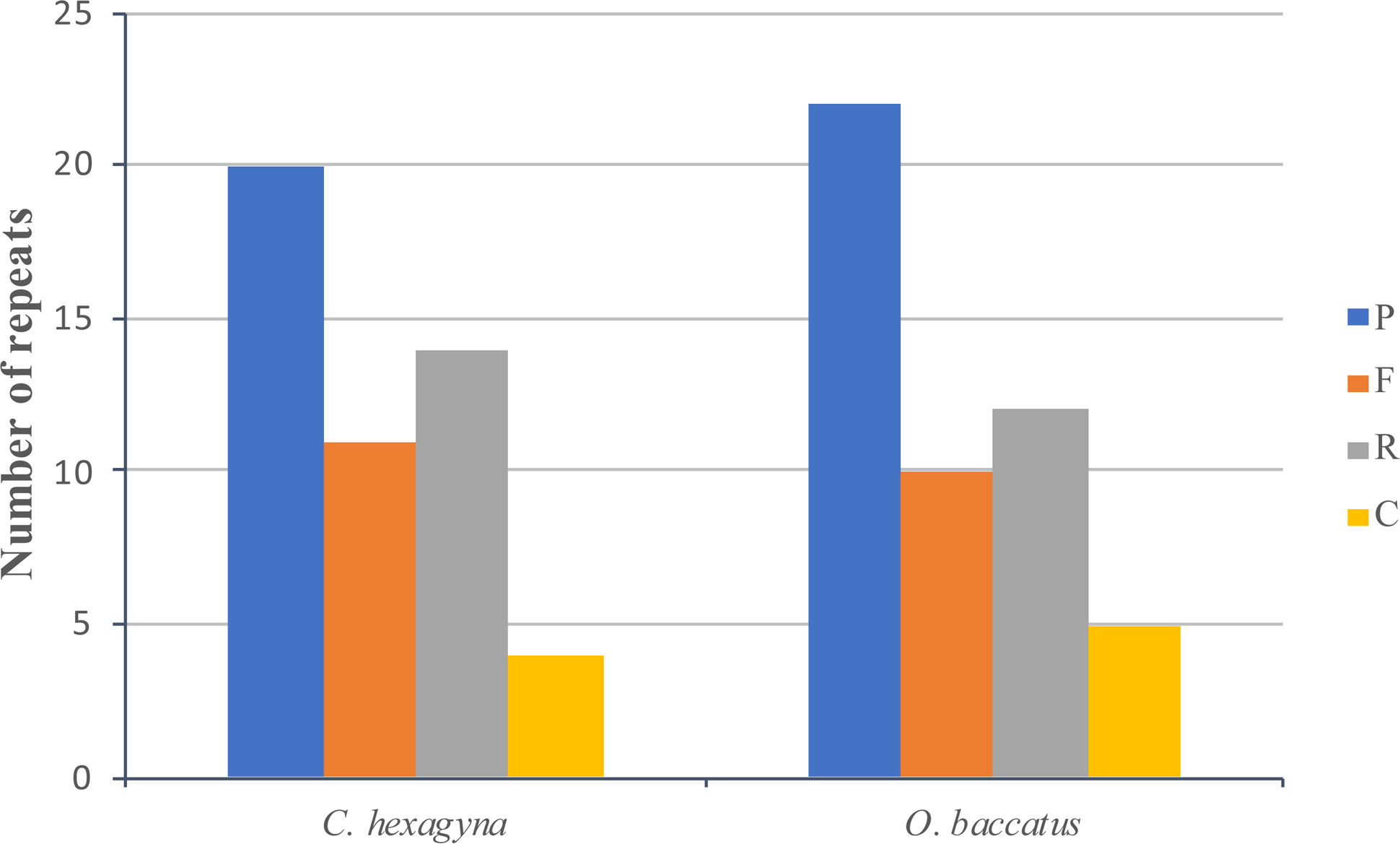
Number of different repeats in the chloroplast genomes of C. hexagyna and O. baccatus. P = palindromic, F = forward, R = reverse and C = complement.
Simple sequence repeats (SSRs)
Short sequence repeats, or microsatellites (SSRs) are dispersed throughout the genome. The chloroplast genome of C. hexagyna contained 256 microsatellites, and there were 255 microsatellites in O. baccatus (S7, S8 Table). In the chloroplast genome of both species, the majority of SSRs were mononucleotides (89.4%), of which the majority were poly A or poly T (Figure 4 and Table 4). Poly A (polyadenine) constituted 38.67% of the mononucleotides in C. hexagyna and 38.43% in O. baccatus, whereas poly T (polythymine) constituted 48.43% of the mononucleotides in C. hexagyna and 49% in O. baccatus. Only three poly C (polycytosine) mononucleotides were present in C. hexagyna (1.17%), with four in O. baccatus (1.56%); there were two poly G (polyguanine) mononucleotides in both species (0.78%). In the genomes of both species, only one dinucleotide (AT/AT) was found in the genome, with one trinucleotide (AAT/ATT), two tetranucleotides (AAAG/CTTT and AAAT/ATTT), one pentanucleotide (AAAAG/CTTTT in C. hexagyna and AAAAT/ATTTT in O. baccatus), and one hexanucleotide (AATAGG/ATTCCT in O. baccatus but none in C. hexagyna) (Figure 4 and Table 4). Microsatellites were more prevalent in the intergenic spacer region in C. hexagyna and O. baccatus (83.59% and 82.35% respectively) than in the coding region (17.96% and 18%, respectively) (Figure 5).
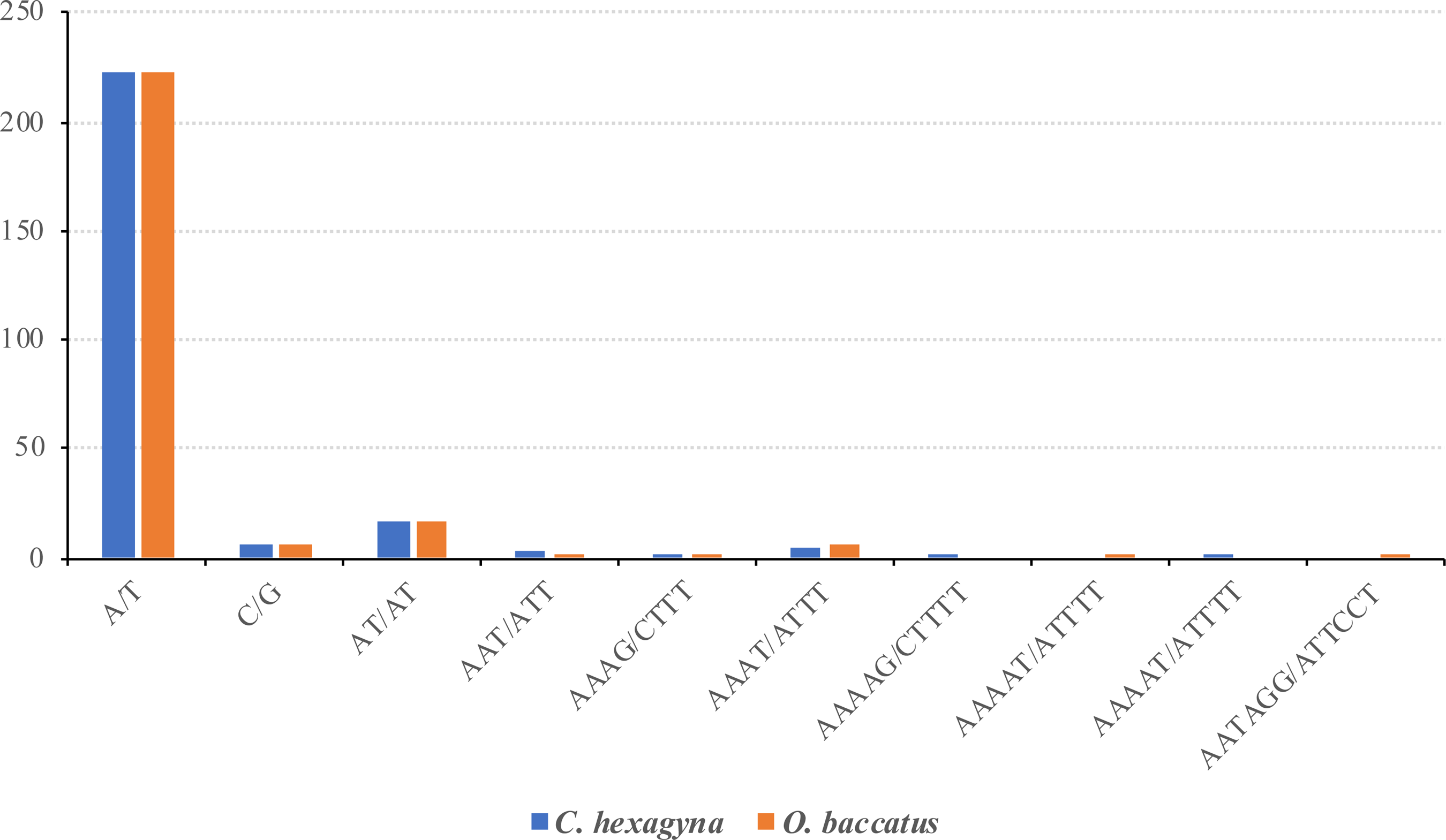
Frequency of different SSR motifs in different repeat types in the C. hexagyna and O. baccatus chloroplast genomes.
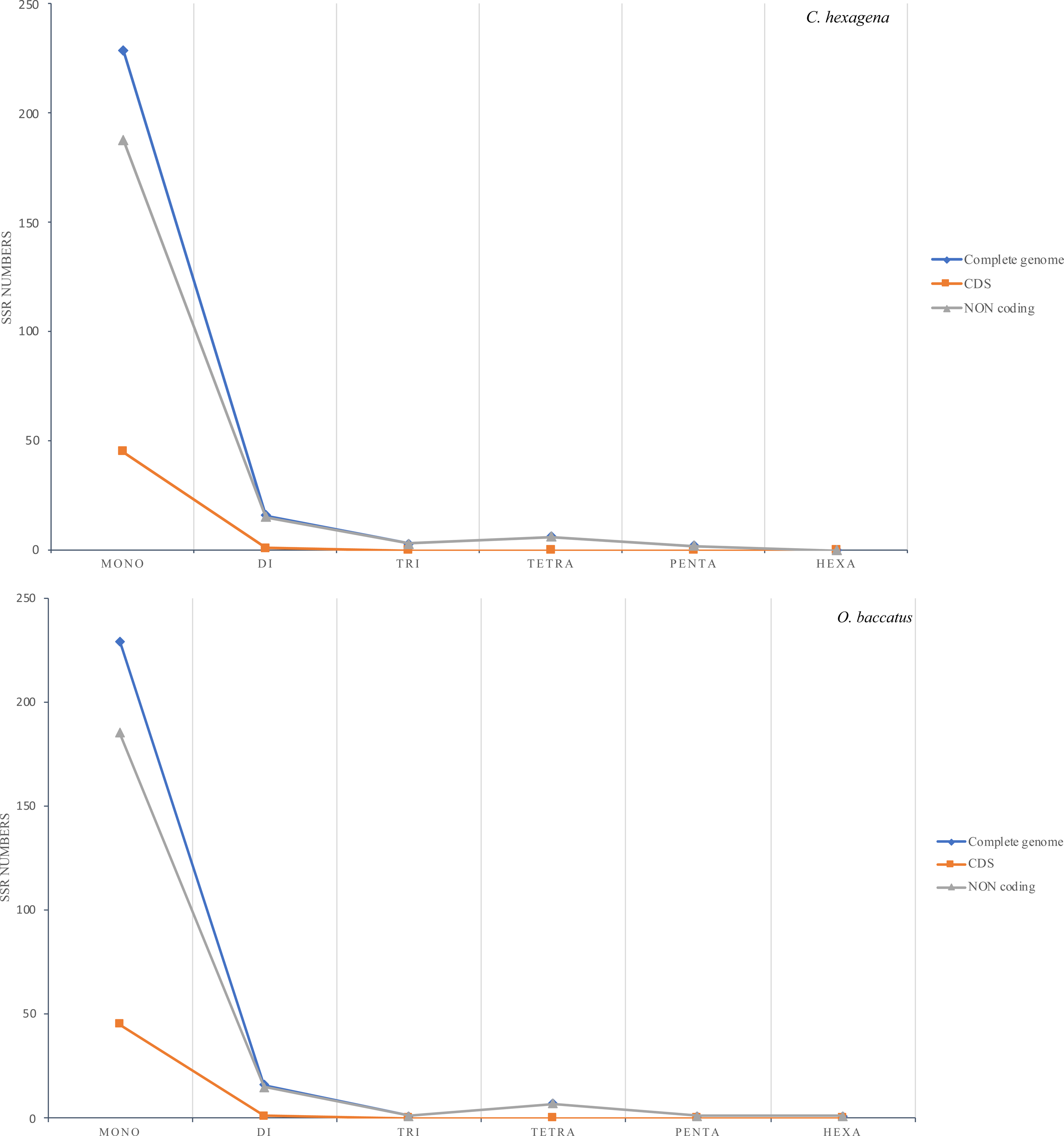
Number of SSR types in the complete genomes, protein-coding regions and noncoding genes in C. hexagyna and O. baccatus.
The SSRs in two chloroplast genomes of Resedaceae.

Comparative analysis
In the current study, the IR-LCS and IR-SSC boundaries of the genomes of the two Resedaceae species were compared. Although the results showed that there were similarities between the cp genomes of both species (Figure 6), O. baccatus had the smallest chloroplast genome (153,380 bp), whereas C. hexagyna had the largest chloroplast genome (154,390 bp). The IR region was 26,526 bp in C. hexagyna and 26,251 bp in O. baccatus. Furthermore, the lengths of the LSC regions were 83,870 bp in C. hexagyna and 83,023 bp in O. baccatus, and those of the SSC regions were 17,468 bp in C. hexagyna and 17,241 bp in O. baccatus. Additionally, comparative analysis of the cp genome of these two Resedaceae species revealed that the location of the rpsl9 gene is between the LSC and IRb regions. The ycf1 gene was located at the boundary of the IRb/SSC regions in C. hexagyna (1051 bp/4225 bp) and O. baccatus (1036 bp/145 bp) and the boundary of the SSC/IRa regions in C. hexagyna (2 bp/45 bp) and O. baccatus (4247 bp/1036 bp). The ndhF was found at the boundary of the IRb/SSC regions, with 43 bp in the IRb region and 2195 bp in the SSC region, in O. baccatus; it was found at the boundary of the SSC/Ira regions, with 2192 bp in the SSC region and 45 bp in the IRa region, in C. hexagyna.

Comparison of the borders of the IR, SSC and LSC regions between the two chloroplast genomes of Resedaceae.
Divergence of protein-coding gene sequence
The rates of synonymous (dS) and nonsynonymous (dN) substitutions and the dN/dS ratio were calculated to detect the selective pressure on 79 protein-coding genes in the plastid genome of two Resedaceae species. The results showed that the dN/dS ratio was less than 1 in all of the paired genes except rpl22 and ycf2 in C. hexagyna vs O. baccatus, with values of 1.14 and 5.57, respectively. The synonymous (dS) subsituation values in all the genes ranged from 0 to 0.12 (Figure 7).

The synonymous (dS) subsituation and dN/dS ratio values of 79 protein-coding genes from two Resedaceae plastid genomes.
Phylogenetic analysis
Phylogenetic relationships based on the Bayesian and maximum parsimony analysis placed all samples into four main clades, and the results matched across the two analyses with strong support for nodes PP and MP, both 1.00 (Figure 8). The two Resedaceae species formed one clade, while the other three families formed in one main clade includes three subclades. The first subclade contained three species of the Capparaceae family. The second subclade contained four Cleomaceae species which was found to be sister to Brassicaceae, while the third clade included eight species from the Brassicaceae family. The phylogenetic tree showed the taxonomic position of the family Resedaceae in the order Brassicales.13,14

Phylogenetic tree reconstruction based on the complete chloroplast genome of sixteen taxa inferred from Bayesian inference (BI) methods showing relationships within resedaceae and other families in brassicales. Numbers in the clade represent posterior probability (PP) values.
Discussion
The complete chloroplast genome has provided much genetic information and many molecular markers as valuable tools to investigate obscure phylogenetic relationships among land plants. 38 The plastid genomes of C. hexagyna and O. baccatus have similar structures to the chloroplast genomes of other angiosperms.35,36,39 This study is the first to report the complete plastid genomes for the genera Caylusea and Ochradenus and for Resedaceae in general.
The plastid genome size of C. hexagyna is 154,390 bp, and that of O. baccatus 153,380 bp (Figure 1). The chloroplast genome sequences of C. hexagyna and O. baccatus had GC contets of 36.49% and 36.42% and AT contents of 63.48% and 63.57%, respectively (Table 1). The GC content in the IR region was 42.2% in both species, which was higher than that of the LSC and SSC regions (Table 1).
There were 113 genes in both genomes, which included 79 protein-coding genes, 30 tRNA genes and four rRNA genes. The IR region contained eight protein-coding genes, seven tRNA genes and four rRNA genes; the LSC region contained 60 protein-coding genes and 22 tRNA genes; and the SSC region contained 12 protein-coding genes and one tRNA gene. Introns are present in several of the protein-coding and tRNA genes in both chloroplast genomes, similar to other chloroplast genomes of flowering plants.35,36,39 Out of the total genes in the cp genomes of both species, 13 genes contained introns, eight were protein-coding genes and five were tRNA genes (Table 3).
The results showed that the genes in the chloroplast genome were encoded by 26,439 codons in C. hexagyna and 26,646 codons in O. baccatus. The most common codons coded for the amino acid leucine, which has been previously stated for several cp genomes of flowering plants. 40 The RNA editing site results revealed that most of the amino acid conversions in the codon positions were serine to leucine, presenting 41 editing sites distributed among 15 protein-coding genes in C. hexagyna and 39 editing sites distributed among 14 protein-coding genes in O. baccatus. The repeat sequences were identified in the chloroplast genomes of C. hexagyna and O. baccatus using default settings, and the long repeat analysis showed 20 and 22 palindromic repeats, 11 and 10 forward repeats, 14 and 12 reverse repeats and 4 and 5 complement repeats, respectively (Figure 3). The length of repeated sequences in the two chloroplast genomes ranged from 10 to 69 bp, which is analogous to the lengths in other angiosperm plants.37,41,42 The majority of SSRs in the cp genomes were mononucleotides, of which most were poly T and A (Figure 4 and Table 4) as in most flowering plant cp genome. 37
This study compared the IR-LCS and IR-SSC boundaries of the two plastid genomes of Resedaceae, and the results showed that O. baccatus had the smallest chloroplast genome. The smallest IR region was in C. hexagyna, and the smallest LSC and SSC regions were in O. baccatus. The results of the rates of the selective pressure among 79 protein-coding genes in the chloroplast genomes of the two Resedaceae species revealed that the dN/dS ratio was less than 1 in all of the paired genes, with a few exceptions (Figure 7). The identical (dS) values in all of the protein-coding genes range from 0 to 0.12 (Figure 7). Phylogenetic relationships based on the Bayesian and maximum parsimony analysis placed all samples into four main clades, where every family was in a separate clade (Figure 8). The phylogenetic tree showed that the family Resedaceae were sister to all other families in the Brassicales.
Conclusion
This current study used the Illumina HiSeq 2500 platform to sequence the complete chloroplast genome of Caylusea hexagyna and Ochradenus baccatus, which provided valuable plastid genomic resources for these plants. We annotated the cp genome for both species; moreover, we identified the base composition, codon usage and RNA editing sites, SSRs and long repeats in these genomes. This study has also provided the first insight into the systematic position of two species (C. hexagyna and O. baccatus) whereby these valuable resources could be used for further phylogenetic and evolutionary relationship studies of the species in the Resedaceae family in order to resolve the taxonomic problems of species such as: Borthwickia, Neothorelia, Stixis, Tirania and Forchhammeria.
Supplemental Material
sj-docx-1-sci-10.1177_00368504211059973 - Supplemental material for The first complete chloroplast genome sequences in Resedaceae: Genome structure and comparative analysis
Supplemental material, sj-docx-1-sci-10.1177_00368504211059973 for The first complete chloroplast genome sequences in Resedaceae: Genome structure and comparative analysis by Dhafer Alzahrani, Enas Albokhari, Abidina Abba and Samaila Yaradua in Science Progress
Footnotes
Authors’ Note
Conceptualization, D.A., E.A. and S.Y.; collected data, D.A and E.A; formal analysis, E.A. and S.Y.; writing—original draft preparation, D.A. and E.A.; writing—review and editing, S.Y. and A.A.; All authors have read and agreed to the published version of the manuscript. The data that support the findings of this study are openly available in the NCBI GenBank at https://![]() , reference number (C. hexagyna MT948187; O. baccatus MT948189).
, reference number (C. hexagyna MT948187; O. baccatus MT948189).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
Author biographies
Dhafer Ahmed M. Alzahrani was born in Saudi Arabia. He obtained his PhD in plant systematics, from the Centre for Plant Diversity and Systematics, School of Biological Sciences, Plant Sciences, University of Reading, United Kingdom, in 2011. He is now an associate professor in the Department of Biological Sciences, Faculty of Sciences, King Abdulaziz University, Jeddah, Saudi Arabia. His specialization includes botany, systematics of flowering plants, and molecular phylogeny. His field of interest includes plant diversity, conservation of plants, flora of Saudi Arabia, and plant ecology. He has published more than 34 research papers in reputable journals and presented about 11 papers in various conferences.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.