
Abstract
Background:
Systemic lupus erythematosus is a common autoimmune disease involving multiple systems. Clinical involvement of the central and peripheral nervous systems is not unusual, but peripheral neuropathy in systemic lupus erythematosus with chronic inflammatory demyelinating polyneuropathy is uncommon. Our study aimed to illustrate the clinical features, diagnosis, and treatment of systemic lupus erythematosus combined with chronic inflammatory demyelinating polyneuropathy, and to aid in the identification of peripheral neuropathy in systemic lupus erythematosus.
Methods:
This article reports a case of systemic lupus erythematosus with onset in pregnancy, with chronic inflammatory demyelinating polyneuropathy as the first manifestation. We then analyze the identification of common peripheral neuropathy in systemic lupus erythematosus in detail, based on a literature review of confirmed cases of systemic lupus erythematosus combined with chronic inflammatory demyelinating polyneuropathy.
Results:
A 34-year-old woman presented progressive muscle weakness and muscular atrophy in the extremities during pregnancy, 3 years previously. At 4 months after onset, she had completely lost the ability to hold objects and walk, and had slight numbness in the limbs, without paresthesia. Her condition was misdiagnosed as “motor neuron disease” at the time. Three years after onset, her condition was revisited because of nephrotic syndrome, and she was diagnosed with nephrotic syndrome and peripheral nerve injury caused by systemic lupus erythematosus. After immunosuppressive treatment with corticosteroids and intravenous cyclophosphamide, her symptoms of muscle weakness were markedly improved. This article summarizes the characteristics of systemic lupus erythematosus combined with chronic inflammatory demyelinating polyneuropathy that have been reported in the literature, from the aspects of morbidity, disease progression, nerve injury, laboratory examinations, and treatment response.
Conclusions:
Our identification of a common peripheral neuropathy in systemic lupus erythematosus will help to improve clinicians’ understanding of various peripheral neuropathies in systemic lupus erythematosus. It will also aid in the early diagnosis and treatment of such patients, thus improving their long-term prognosis.
Keywords
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease caused by multiple autoantibodies, involving multiple systems. It is most common in young women and has various clinical manifestations.1,2 Although the skin, joints, blood system, and kidneys are the most commonly involved organs in SLE,3–6 the nervous system is also often affected, and the central nervous system is most commonly involved (e.g. lupus encephalopathy).7,8 In contrast, symptomatic peripheral nervous system involvement is relatively uncommon in SLE,8,9 and SLE with the first symptoms involving the motor system is very rare.
Methods
We report a case of SLE with progressive myasthenia, accompanied by muscular atrophy as the first symptom, which was misdiagnosed as “motor neuron disease.” We then review the relevant literature to provide suggestions for the clinical diagnosis and treatment of this rare SLE. Informed consent was obtained from the participant included in the study.
Results
Case report
A 34-year-old female patient was admitted to the hospital in 2019, because of “limb weakness with muscle atrophy for more than 3 years and edema for more than 1 month.” Three years previously, the patient, who was at 16 gestational weeks, had felt limb weakness for no obvious, predisposing cause and her fatigue gradually increased. At 28 gestational weeks, her muscle weakness progressed to an inability to walk or hold objects (Figure 1A), and was accompanied by muscle atrophy without any obvious sensory abnormalities except slight numbness. The patient then went to the neurology department of a local hospital and received a routine blood examination and urine analysis, but no obvious abnormalities were noted. The patient was diagnosed with “motor neuron disease.” However, she did not continue the examination or treatment because of her pregnancy. After the delivery at 36 gestational weeks, she was prescribed traditional Chinese medicine (the ingredient in this medicine is unknown). At 3 – 4 months after the delivery, some of her symptoms of weakness were alleviated compared with during pregnancy. She was able to partially spread her fingers, but her standing and walking abilities remained the same as before, and the traditional Chinese medicine was suspended. She was able to stand with assistance at 1 year after the delivery and was able to walk with a walker 2 years after the delivery. However, 1 month before the present report, edema occurred in the patient's face and lower limbs with no inducing factors, and this was accompanied by dizziness, cough, and sputum. She also suffered from occasional lumbar pain and foam in the urine, but did not have fever, chills, gross hematuria, dry mouth, dry eyes, light allergy, oral ulcers, or joint pain. The urine analysis at a local hospital showed urine protein ( + + + ); urine occult blood (±); urine glucose (−); and urine protein quantitation in 24 h: 3.3 g/24 h. She also had blood albumin: 15.1 g/L; creatinine: 43 μmol/L; total cholesterol: 6.74 mmol/L; triglycerides: 3.08 mmol/L; blood calcium: 1.84 mmol/L. The patient was diagnosed with “nephrotic syndrome” and treated with kidney protection (Bailing Capsule, Fermentative Cordycepis Fungal Powder) and a calcium and albumin supplement, but the swelling symptom continued without change. She was then admitted to our hospital for further evaluation and treatment, with a diagnosis of “nephrotic syndrome.”
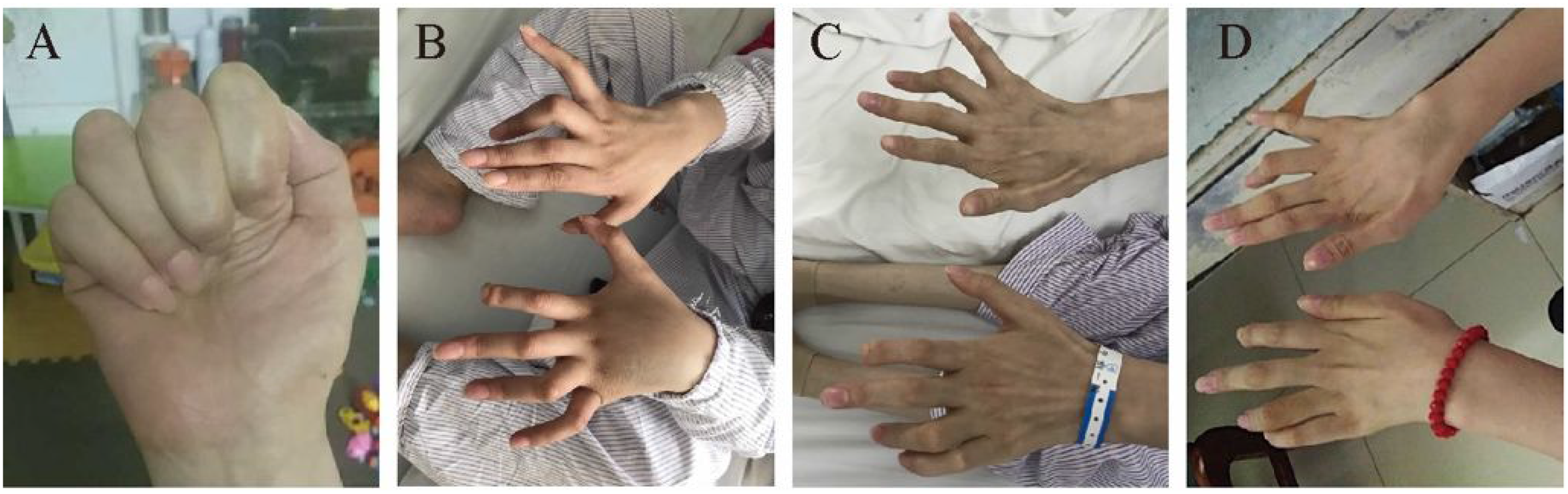
Photos of the patient's hands: (A) the patient simulating the state in which her fingers were unable to be opened, which occurred during the 7th month of pregnancy; (B) the patient's hands during her first hospitalization with renal syndrome; (C) the patient's hands when she returned to the hospital for the second time; (D) current photo of the patient's hands.
Diagnosis and treatment
Physical examination showed her bilateral eyelids were slightly swollen. Her heart rate and her cardiac rhythm were regular. Blood pressure was 144/96 mmHg. Auscultation of the bilateral lungs was normal. The abdomen was soft and shifting dullness was positive. There is severe pitting edema from bilateral feet to thighs. Neurological examination revealed no abnormalities of the cranial nerve. The muscle tone of all limbs was low. The proximal muscle strength of the upper limbs was grade 0, while the distal strength was grade IV. The patient's fingers had deformities and her hands were atrophic (Figure 1B). The proximal muscle strength of the lower limbs was grade IV-V, while the distal strength was grade 0. She exhibited bilateral foot drop, but sensation was normal and symmetrical. Tendon reflexes in all limbs were non-existent, and pathological reflexes were negative.
After admission, the patient accepted to undergo clinical examinations. Routine blood examination, alanine aminotransferase, and aspartate aminotransferase showed normal. She had severe hypoalbuminemia (14 g/L) and hypogammaglobulinemia (19 g/L). Serum creatinine was normal (46 mmol/L). Urine analysis showed urine protein ( + +), microscopic hematuria: 5/µL, and urine protein quantitation in 24 h: 24 g/24 h. The erythrocyte sedimentation rate was 29 mm/h and the C-reactive protein level was 1.62 mg/L. The autoantibody and immune markers tests are shown in Table 1.
Summary of antibodies and immune markers.

ANA: antinuclear antibodies; anti-dsDNA: anti-double-stranded DNA antibody; SSA/SSB: Sjögren syndrome A/B; SM: smooth muscle; RNP: ribonucleoprotein; ACA: anti-centromere autoantibody.
Doppler echocardiography showed left atrial enlargement and mild pericardial effusion. Ultrasound of the hand joints showed no obvious abnormalities in the joints or tendons of either hand. The bilateral median nerves in the carpal tunnel exhibited no abnormalities, but they were diffusely thicker from the forearm to the brachial plexus (Supplemental Figure 1). The inner structures of bilateral median nerves were clear and had good continuity. Nerve conduction studies (Table 2) demonstrated that the distal latencies of nerve motor conduction were prolonged and the motor potential amplitudes were reduced in the left axillary nerve, right median nerve, and right ulnar nerve. Motor action potentials were unable to be elicited in the left median and ulnar nerves and the right common peroneal nerve, as well as in the bilateral median and ulnar sensory nerves. The latency of the F wave was prolonged in the right median nerve. Electromyography (EMG) showed abnormal spontaneous potentials in the left anterior tibialis anterior muscle during the resting state. The left deltoid muscle and extensor digitorum communis had widened mean durations and high amplitudes in the motor units. The maximum recruitment of these motor units showed interference patterns. The patient's left anterior tibialis muscle was too weak to contract (Table 3 and Figure 2). All of these dysfunctions indicated that the peripheral nerves of all limbs were severely damaged, and suggesting demyelination and axonal damage. EMG indicated neurogenic injury. The clinical diagnoses were: (1) SLE, lupus nephritis, and nephrotic syndrome; and (2) immune-related peripheral neuropathy (most likely related to SLE). Since her diagnosis of SLE was very clear and she was at a prominent hypercoagulable state, anticoagulant treatment was prescribed and kidney biopsy was suspended. The patient was treated with lipid-lowering therapy, kidney protection, and calcium and albumin supplement. She was given 5–10 g intravenous immunoglobulin for 7 days as well as mecobalamin for neural protection. She also received 250 mg methylprednisolone daily for 3 days and 125 mg methylprednisolone for 1 day, and then 40 mg methylprednisolone via intravenous drip every day as treatment. In addition, she was administered cyclosporine for immunosuppressive therapy, at a dose of 75 mg every morning and 50 mg every night. Because of severe edema and chest discomfort, we also administered pure ultrafiltration treatment, which alleviated the patient's edema considerably. After 10 days of comprehensive treatment, her muscle strength improved and she was able to spread most of her fingers. She was able to hold objects weighing 10 kg and walked with a walker. Because the local hospital did not have the capacity to monitor cyclosporine concentrations, and she was skinny and malnourished at the very time, and our experience showed that Chinese are more sensitive to MMF than the Westerners, we start mycophenolate mofetil (MMF, CellCept) from a very low dosage (0.25 g q 12 h) along with 40 mg oral methylprednisolone every morning as discharge medication. Hydroxychloroquine had been prescribed throughout the course.

EMG images of the patient: (A) EMG MUPs of left deltoideus; (B) EMG MUPs of left extensor digitorum communis.
Nerve conduction studies of the patient.

ADM: adductor digiti minimi muscle of hand; APB: abductor pollicis brevis; AH: abductor hallucis; EDB: extensor digitorum brevis. Wrist – ADM: elbow-wrist, wrist – APB: ankle-AH, Erb-Deltoid muscle: ankle-EDB, knee-ankle refer to placement of stimulation electrode to recording electrode, respectively. “–” refers to no elicited motor action potentials. L.: left; R.: right; ms = millisecond, m/s = meter per second, mv = millivolt. Reference values are shown in parenthesis, as upper/lower limit of normal or mean ± SD, according to Buschbacher et al. 10
EMG results of the patient.
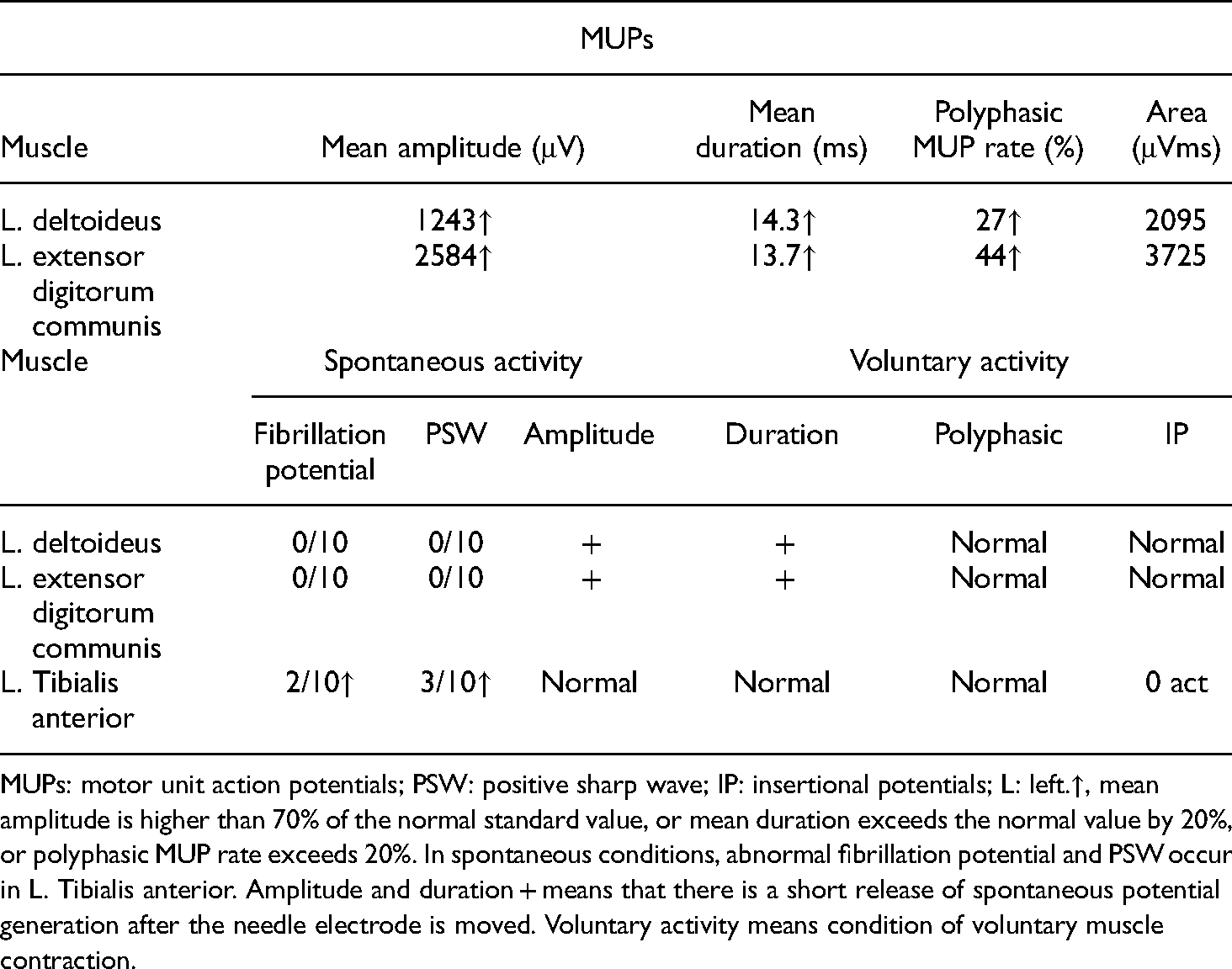
MUPs: motor unit action potentials; PSW: positive sharp wave; IP: insertional potentials; L: left.↑, mean amplitude is higher than 70% of the normal standard value, or mean duration exceeds the normal value by 20%, or polyphasic MUP rate exceeds 20%. In spontaneous conditions, abnormal fibrillation potential and PSW occur in L. Tibialis anterior. Amplitude and duration + means that there is a short release of spontaneous potential generation after the needle electrode is moved. Voluntary activity means condition of voluntary muscle contraction.
Follow-up and prognosis
The patient went back to her hometown after being discharged from our hospital. She took oral prednisone at an equivalent dose to methylprednisolone, and after 2 months she began to reduce the dose by 5 mg every 2 weeks. She stopped taking mycophenolate mofetil (0.25 g every 12 h) 2 months after being discharged from hospital because MMF was not available at her hometown. Prednisone and hydroxychloroquine were continued. When the prednisone was slowly reduced to 20 mg every morning (6 months after being discharged), the patient visited our hospital for a second visit. At this visit, the muscle strength in her hands was much better than before (Figure 1C). She was able to eat food by herself using chopsticks, hold objects weighing 15 kg, and walk with a crutch according to her description. For her convenience, we changed her treatment to 0.6 g cyclophosphamide per month via intravenous drip, and continued the oral prednisone at 20 mg every morning. At a follow-up visit during the 17th month after the patient's first admission to our hospital, the cyclophosphamide cumulative dose was 4.2 g (seven administrations in total), and the corticosteroid was reduced to 7.5 mg every morning. She went to her local hospital for re-examination. Urine protein quantitation in 24 h was 0.588 g/24 h, and blood albumin was 32 g/L. The edema symptoms had disappeared, and the limb weakness symptoms had also made favorable progress. Figure 1D shows her current photo of her hands. She was able to eat foods using chopsticks and dress by herself, hold objects weighing 20–25 kg, and walk slowly with a crutch, according to her description. She was also able take care of herself in daily life. During the treatment, there were no obvious side effects of the corticosteroid or immunosuppressive treatments.
Literature review of the confirmed cases of SLE combined with chronic inflammatory demyelinating multiple neuropathy (CIDP)
SLE combined with CIDP is uncommon and is easily misdiagnosed or missed. To gain a better understanding of SLE and CIDP so that patients can be diagnosed earlier, we searched PubMed using the following search terms: “CIDP, chronic inflammatory demyelinating polyneuropathy, SLE, systemic lupus erythematosus.” Case reports and case series in English-language articles until January 2020 were considered. We then summarized the confirmed cases in the literature, as shown in Table 4.
Summary of the literature on systemic lupus erythematosus with chronic inflammatory demyelinating disease.

cANCA: Anti-neutrophil cytoplasmic antibodies; lv: intravenous; IVIG: intravenous immunoglobulin; MRI: magnetic resonance imaging.
Clinical manifestations
Patients with SLE combined with CIDP were found at all ages, and no trend was observed of primary CIDP increasing with age. In addition, SLE combined with CIDP has been reported in both men and women, and in a range of races. The onset time is mostly longer than 2 months, but several cases have peaked at 3–4 weeks, manifesting as acute-onset CIDP. The affected nerves in SLE combined with CIDP mainly involve the limbs and facial nerves, and the disease presents a pattern of slow progression. Motor nerve damage mainly manifests as facial and limb weakness, and walking disorders; however, in severe cases, patients become bedridden. Sensory nerve damage is mainly manifested as dysesthesia and numbness of the extremities, as well as impaired vibration senses. SLE with CIDP patients rarely experience autonomic nerve involvement, but they all have reduced or absent tendon reflexes. Most cases are accompanied by skin rash, proteinuria, and other SLE-related systemic damage. Five reported cases had cranial nerve damage, leading to a variety of clinical manifestations, such as facial weakness, numbness, loss of taste, limb tremor, dysarthria, and other manifestations corresponding to different affected nerves. The majority of patients with SLE have characteristic antibody positivity and hypocomplementemia. Their magnetic resonance imaging results are normal, and cerebrospinal fluid examinations show a characteristic protein–cell separation (increased protein levels with normal cell numbers). Furthermore, most nerve conduction studies have reported reduced amplitude and velocity of nerve potentials, prolonged distal latencies, and abnormal F waves. Electrical nerve examinations show clear sensory and motor nerve damage.
Treatment and prognosis
In patients with SLE combined with CIDP, the treatments used have been different. All cases received steroid therapy. However, some patients received steroid therapy alone, while other patients received steroid therapy combined with intravenous immunoglobulin therapy or immunosuppression. Considerable individual differences in treatment response and prognosis have been reported. Some patients had rapid alleviation of neurological symptoms after steroid treatment alone, while other patients obtained long-term relief after combined treatment; moreover, the degree of obtained relief was often different.
Discussion
This article reports a rare case of SLE in a middle-aged woman, which occurred during pregnancy. This patient met both the 1997 American College of Rheumatology (ACR) criteria for SLE 18 (antinuclear antibody positive, double-stranded DNA antibody positive, pericardial effusion, large amounts of proteinuria) and the diagnostic criteria of the European Society of Neurology and the Federation of Peripheral Neurological Societies for CIDP 19 (chronically progressive, stepwise, or recurrent symmetric proximal and distal limb weakness, with sensory dysfunction of all extremities that developed over 2 months, with reduced or absent tendon reflexes; not caused by infections, drugs, inheritance, or other reasons; electrophysiological examination is consistent with peripheral nerve demyelination changes). We therefore considered our patient's diagnosis as SLE combined with CIDP.
Clinically, SLE patients with nerve damage are very common, 20 but this damage is mostly related to the central nervous system, and usually manifests as epilepsy, lupus headache, acute confusion, anxiety, cognitive function decline, emotional disorders, or mental disease. Its incidence rate is 37–90%.21–23 SLE with peripheral nervous system involvement is also common. Approximately 21–42% of all SLE patients have abnormalities in nerve conduction tests, 24 but the symptomatic SLE-related peripheral neuropathy incidence rate is very low in the clinic, at only 4.9–7.6% of SLE patients with neuropsychiatric manifestations.8,25 The ACR defines seven types of peripheral neuropathy: acute inflammatory demyelinating polyradiculoneuropathy (Guillain–Barré syndrome (GBS)), autonomic disorder, mononeuropathy, myasthenia gravis, cranial neuropathy, plexopathy, and polyneuropathy 26 ; however, CIDP is not included. The most common type is symmetrical polyneuropathies, 27 of which distal sensorimotor axonal polyneuropathies is the most common form. 28 Distal sensorimotor axonal polyneuropathies generally manifests as distal symmetric polyneuropathy, with two main characteristics: pain and progression, mostly starting from the distal end (e.g. on the foot) and expanding proximally, although electrophysiological examinations reveal asymmetry in nerve conduction. 24
When muscle weakness is the first symptom in SLE, as in our patient, special attention should be paid to the identification of motor neuron disease. Motor neuron disease is a group of progressive degenerative disorders of unknown etiology. 29 This disease selectively invades the motor system or a certain part of the motor system. The range of lesions includes those of the spinal anterior horn cells, cranial nerve motor nucleus, cortical pyramidal tract, and cortical medullary tract. It involves the upper motor neuron and/or the lower motor neuron. The clinical manifestations of motor neuron disease have the characteristics of lower motor neuron damage, relative muscle atrophy and weakness, and medullary palsy, and/or damage to the upper motor neurons (pyramidal tracts). Limb weakness gradually progress. In contrast, the sensory system generally remains intact.29,30 Although lower motor nerve damage was the main manifestation of our patient in the present report, nerve electrophysiological examinations showed severe sensory nerve damage, which is vital for the early differential diagnosis of motor neuron disease. In addition, her muscle weakness had a characteristic progressive exacerbation and progressed most severely when she went to the local hospital in her seventh month of pregnancy. However, it was alleviated after her baby was delivered, which does not reflect the motor neuron disease pathogenesis of irreversible progression but does reflect the repeated illness states of CIPD and SLE. Regrettably, we did not obtain her early routine blood and urine test results, and the patient did not undergo autoantibody-related tests during her two visits to the local hospital. It was therefore unclear whether her kidneys or blood system were already involved with SLE during her early hospital visits.
This patient's condition also needed to be differentiated from generalized myasthenia gravis. Generalized myasthenia gravis is an autoimmune disease that is characterized by motor conduction dysfunction caused by autoantibodies destroying postsynaptic acetylcholine receptors at nerve-muscle junctions. The clinical manifestations of this disease are skeletal muscle volatility weakness and intolerance to fatigue, which worsen after activity and are alleviated after rest. In severe cases, flaccid paralysis can occur. For patients with this disease, the neostigmine test is positive, electrophysiological examination shows that the low frequency of repetitive nerve stimulation decreases by >15%, the cerebrospinal fluid examination is normal, and acetylcholine receptor antibodies can be detected in most patients.31–33 In our patient, the muscle weakness in the early stage of the disease increased progressively, and there was no fluctuation in the morning or evening. The patient's electrophysiological examination revealed that the motor and sensory nerves were severely damaged, which likely excludes generalized myasthenia gravis.
Patients with diabetes may also have peripheral nerve damage, 34 which usually manifests as walking difficulty. The main manifestation of diabetic vascular neuropathy is plexus neuropathy. The typical symptom involves an acute attack of sharp pain on one side of the thigh, which gradually affects the entire leg, with weakness that is often severe enough to require the use of a walker. 35 Clinical examination and EMG usually show nerve root damage, which is often accompanied by autonomic dysfunction and weight loss. 36 However, our patient had no history of diabetes and no pain, so this disease was excluded.
CIDP is an acquired, immune-mediated demyelinating nerve injury disease. Typical CIDP manifests as motor-based symmetric peripheral neuropathy that causes proximal and distal weakness. It also causes sensory disturbances, which usually have a greater impact on vibration and proprioception than on pain and temperature senses. The course of this disease is progressive or recurrent. 9 Typical changes in demyelination, which can be observed in electrophysiological examinations, are essential for the diagnosis of CIDP. When demyelination (i.e. myelin being destroyed) occurs, it takes a longer time for the drive current to activate the next node to threshold, resulting in slower internode conduction and decreased conduction velocity along the nerve segment. 37 Because of the complex electrophysiological expression of demyelination, there is currently no gold standard for its diagnosis. According to the European Federation of Neurological Societies/Peripheral Nerve Society Guideline, 38 the main manifestation of CIDP nerve conduction study (NCS) includes motor distal latency prolongation, and the extension of the F-wave latency. In our case report, NCS shows that the motor conduction of the left axillary nerve and the right median and ulnar nerves distal latency are prolonged. The F wave latency of the right median nerve is prolonged. At the same time, the EMG shows neurogenic injury, which is in line with the electrophysiological characteristics of CIDP. However, although the Academy of Neurology states that demyelination requires a conduction velocity slowdown or conduction block, most CIDP patients fail to meet this standard, either because insufficient fibers are affected when the proximal demyelination is not affected by distal stimulation, or because the secondary axonal degeneration is too severe to be able to observe the conduction slowdown. 37
Although more men have primary CIDP onset, more female patients are affected by SLE with CIDP, which may be related to the relatively high incidence of SLE in young women. A recent review observed that patients with CIDP and SLE are predominantly female. 39 There is currently no report on whether the incidence of CIDP combined with SLE is related to race. CIDP complicated with SLE during pregnancy has not been seen in previously reported cases. This case is the first report. There is one report that studied the onset and recurrence of primary CIDP during pregnancy. It is believed that in some women, pregnancy may lead to typical CIDP, with a higher risk of CIDP recurrence. 40 However, more case studies are still needed to explore. Some CIDP cases with acute onset need to be differentiated from acute inflammatory demyelinating neuropathies (such as GBS). Because both CIDP and GBS are demyelinating nerve injuries, the results of electrophysiological examinations cannot be distinguished; however, the clinical processes of these two diseases are different. GBS often involves respiratory muscles and requires assisted ventilation treatment, and its progression usually peaks within 4 weeks. In contrast, CIDP patients seldom need assisted ventilation, and the condition is prolonged or relapses, so the course of CIPD is longer than that of GBS. 41 Although both motor and sensory nerves are involved in SLE with CIDP, the symptoms of muscle weakness are more prominent, similar to those of primary CIDP. In addition, some patients have no obvious sensory abnormalities, and may not even have abnormalities in vibratory sensation or proprioception by physical examination, with only electrical nerve examinations showing sensory nerve damage. Both proximal and distal muscle groups can be involved in primary CIDP patients, and they are not generally characterized by ascending from distal to proximal. However, some SLE with CIDP patients may have axonal damage caused by simultaneous vasculitis, and their electrophysiological examinations show markedly reduced amplitudes of motor potentials at an early stage. It remains to be identified whether decreased motor potential amplitudes in the later stages of the disease represent primary axonal degeneration or secondary axonal degeneration caused by severe demyelination. A nerve biopsy is unnecessary for a diagnosis of CIDP, because CIDP mainly involves motor nerves, while a biopsy is mainly based on sensory nerves. Nonetheless, if the pathology result shows demyelination changes, it can be used as the basis for a definite diagnosis.
The pathogenesis of CIDP remains unclear. It is currently believed that both cellular and humoral immunity are involved in its pathogenesis. 42 Approximately 6–10% of patients have antibodies against neurofascin and contactin, and these antibodies are likely to be pathogenic.43,44 Studies have also captured the process of macrophages through the basement membrane peeling off myelin flakes, combined with photos of macrophages rich in myelin fragments, suggesting that macrophages induce the demyelination process.45,46 However, most clinical nerve biopsy specimens do not show a large number of infiltrating macrophages.
To date, there have been no large studies or prospective controlled studies on the treatment of peripheral neuropathy in SLE. For SLE treatment, the 2020 Chinese SLE Diagnosis and Treatment Guidelines are as follows 47 : (1) Use hydroxychloroquine as the basic treatment; for severe neuropsychiatric lupus, glucocorticoid pulse therapy can be performed first and (2) Comprehensively consider the patient's clinical manifestations, fertility requirements, drug safety, cost factors, and other factors, and choose the appropriate immunosuppressive agent. Controlled trials of CIDP have shown that corticosteroids, plasma exchange, and intravenous immunoglobulin therapy are effective treatments.48–50 In addition, there have been uncontrolled studies showing that immunosuppressants, such as cyclosporin, interferon α, azathioprine, and cyclophosphamide, are effective. 12 However, there have also been reports that high-dose intravenous immunoglobulin treatments are not effective for SLE with CIDP patients; these patients may require combined immunosuppressant therapy.14,16 Studies on the efficacy of MMF in the treatment of CIDP have found that MMF can relieve the nerve damage of primary CIDP, and reduce the doses and frequency of corticosteroid and intravenous immunoglobulin treatment. 51 Moreover, increasing studies have found that MMF can alleviate SLE, but there are no reports of MMF treatment in cases of CIDP combined with SLE.52,53 In the present report, our patient was treated with corticosteroids combined with intravenous cyclophosphamide, with marked effects. Not only was her nephrotic syndrome relieved, but her symptoms of muscle weakness also improved considerably. For patients with fertility requirements, it has been reported that rituximab combined with adequate corticosteroid therapy can also achieve good results. 54 The present review of cases suggests that patients with SLE and CIDP respond well to corticosteroids and immunosuppressive therapy and that the early damage is reversible. The timely diagnosis and treatment of patients is therefore essential, and can significantly improve patient prognosis.
Conclusions
SLE is a chronic disease with various manifestations and has a protracted disease course. Its wide range of symptoms may cause patients to visit different hospital departments. SLE combined with CIDP is rare in clinical practice. We compared the characteristics of different peripheral neuropathies in SLE, which will enhance clinicians’ awareness of the disease, and especially of its rare symptoms. Our review is conducive to the timely diagnosis and treatment of the disease and will improve the prognosis of patients. In addition, it is vital to improve and perfect each patient's basic routine blood, urine, biochemical, and immune-related tests, to detect abnormalities and carefully analyze their causes so that common diseases that start with rare symptoms can be diagnosed in a timely manner.
Supplemental Material
sj-docx-1-sci-10.1177_00368504211050276 - Supplemental material for Progressive muscle weakness and amyotrophy during pregnancy as the first manifestation of systemic lupus erythematosus: A case report and review of literature
Supplemental material, sj-docx-1-sci-10.1177_00368504211050276 for Progressive muscle weakness and amyotrophy during pregnancy as the first manifestation of systemic lupus erythematosus: A case report and review of literature by Liu Wang, Dan Wang, Yuyi Ruan, Xionghui Chen, Wei Chen, Zhijian Li and Xin Wang in Science Progress
Footnotes
Acknowledgements
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Chinese National Key Technology R and D Program, Ministry of Science and Technology [grant numbers 2017YFC0907601, 2017YFC0907602, 2017YFC0907603, 2016YFC0906100, 2016YFC0906101]; Guangdong Natural Science Foundation [grant number 2020A1515010241]; the Key Laboratory of Nephrology, Guangdong Province [grant number 2020B1212060028]; and the Key Laboratory of National Health Commission [grant number 2002B60118]; Guangdong Basic and Applied Basic Research Foundation [grant number 2019A1515010992]; Science and Technology Program of Guangzhou, China [grant number 201804020049].
Supplemental material
Supplemental material for this article is available online.
Author biographies
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.