
Abstract
Objectives:
Swamp eel is one model species for sexual reversion and an aquaculture fish in China. One local strain with deep yellow and big spots of Monopterus albus has been selected for consecutive selective breeding. The objectives of this study were characterizing the Simple Sequence Repeats (SSRs) of M. albus in the assembled genome obtained recently, and developing polymorphic SSRs for future breeding programs.
Methods:
The genome wide SSRs were mined by using MISA software, and their types and genomic distribution patterns were investigated. Based on the available flanking sequences, primer pairs were batched developed, and Polymorphic SSRs were identified by using Polymorphic SSR Retrieval tool. The obtained polymorphic SSRs were validated by using e-PCR and capillary electrophoresis, then they were used to investigate genetic diversity of one breeding population.
Results:
A total of 364,802 SSRs were identified in assembled M. albus genome. The total length, density and frequency of SSRs were 8,204,641 bp, 10,259 bp/Mb, and 456.16 loci/Mb, respectively. Mononucleotide repeats were predominant among SSRs (33.33%), and AC and AAT repeats were the most abundant di- and tri-nucleotide repeats motifs. A total of 287,189 primer pairs were designed, and a high-density physical map was constructed (359.11 markers per Mb). A total of 871 polymorphic SSRs were identified, and 38 SSRs of 101 randomly selected ones were validated by using e-PCR and capillary electrophoresis. Using these 38 polymorphic SSRs, 201 alleles were detected and genetic diversity level (Na, PIC, HO, and He) was evaluated.
Conclusions:
The genome-wide SSRs and newly developed SSR markers will provide a useful tool for genetic mapping, diversity analysis studies in swamp eel in the future. The high level of genetic diversity (Na = 5.29, PIC = 0.5068, HO = 0.4665, He = 0.5525) but excess of homozygotes (FIS = 0.155) in one breeding population provide baseline information for future breeding program.
Introduction
Simple sequence repeats (SSRs) or microsatellites are a group of DNA sequences consisting of tandemly repeated units (1–6 bp), and they are present in both protein coding and non-coding regions of the genome. 1 Owing to the characteristics of simplicity, abundance, ubiquity, co-dominance, and highly polymorphism, SSRs have become a common tool in fish and aquaculture, including genetics,1,2 genomics, 3 characterization of genetic stocks, 4 marker-assisted selection, 5 parentage determination, 6 and linkage analysis and quantitative trait loci (QTL) mapping. 7 Recently, genome-wide identification and development of SSR markers have been successfully performed in various fish, such as spotted sea bass (Lateolabrax maculatus), 8 common carp, 9 and spotted scat (Scatophagus argus), 10 and many marine animals. 11
Swamp eel (Monopterus albus) has been a model species for studying sexual reversal,12–14 and it is becoming an important freshwater aquaculture fish in China 15 and South-East Asia due to its flavor and nutrition.16,17 As different genetic lineages and multiple local strains of swamp eel are present in China,18,19 one local strain with deep yellow and big spots that mainly distributed in Jianghan plain has been selected for consecutive selective breeding due to superiority in growth rate and fecundity. Marker-assisted selection (MAS) would provide great help for accelerating the breeding process in fish, 20 and SSRs have been extensively applied in such breeding programs in many aquaculture fishes.7,21 However, only limited SSRs had been developed22–25 in swamp eel. Although genome-wide SSRs have been characterized swamp eel, only a total of 99,293 loci were identified, and few of them have been validated in this species. 26
Here, based on the genome assembly of the deep yellow and big spots strain of swamp eel in our previous work, 27 we characterized the density, type, and distribution of SSRs motifs, and developed polymorphic genomic SSR markers in this work. This study contributes to increasing the number of molecular markers available for genetic studies in M. albus, which will allow the development of breeding programs in the future.
Materials and methods
Animal care
The experimental procedure was approved by the Animal Experimental Ethical Inspection of Laboratory Animal Centre, Yangtze River Fisheries Research Institute, Chinese Academy of Fishery Sciences (ID Number: 2020-THF-01).
Identification of genome wide SSRs
The reference genome sequences of M. albus (with deep yellow and big spots) were downloaded from Genome Sequence Archive in National Genomics Data Center under accession number CRA003062. 27 Perl scripts from MISA were used to perform SSR identification with the default parameters (http://pgrc.ipk-gatersleben.de/misa/). 28 Initially SSRs of 1–6 nucleotides motifs were identified with the minimum repeat unit defined as 10 for mononucleotides, 6 for dinucleotides, 5 for trinucleotides, 4 for tetra-nucleotides, and 3 each for penta- and hexa-nucleotides. Compound SSRs were defined as those with an interval less than 100-nt between two repeat motifs. For simplicity, those repeats with unit patterns being circular permutations and reverse complements were grouped as one type. 29 For instance, AGG denotes AGG, GGA, GAG, CCT, CTC, and TCC in different reading frames or on the complementary strand. Relative frequency (SSRs per megabase pair (Mbp)) and relative density (SSRs in base pairs (bp) per Mb) were used to help conduct comparisons between different repeat types. 30
Identified SSRs were assigned to genomic compartments using the BEDtools intersect tool v2.29.0. 31 The sequences and coordinates of gene models, exons, coding sequences (CDSs), and intronic and intergenic regions for the swamp eel genomes were determined according to the positions in the genome annotation files in GFF format reported previously. 27
Primer design for genome wide SSRs
To design primers for identified SSRs, two interface Perl script modules (p3_in.pl and p3_out.pl) were used to interchange data between MISA 28 and the primer designing software Primer 3. 32 The primer design criteria were as follows: primer length was between 18 and 27 bp with 20 bp as optimum, melting temperature was from 57°C to 63°C, GC content ranged from 30% to 70%, and product size was from 100 to 280 bp. An R package (http://www.R-project.org), Cmplot, was used to draw the high-density physical map of the newly developed SSR markers.
Screen and validation of polymorphic SSRs
Based on our assembled genome and the available transcriptomes of swamp eel, 33 Polymorphic SSR retrieval tool (PSR) 34 were deployed to detect SSR length polymorphisms of perfect repeats. Allele number was taken as a measurement to evaluate the polymorphism of each type of motif among different samples.
We only choose polymorphic SSRs with tri- or more nucleotide repeat motifs to validate in the following analysis. Initially, primers were designed and validated their amplification efficiency by using electronic PCR (e-PCR) (version 2.3.12) over our sequenced genome data as templates. Then a total of 101 newly developed primer pairs for SSRs were randomly selected and tested for polymorphism with six individuals by capillary electrophoresis of amplicons. The 5′ ends of the forward primer of each primer pair was labeled with FAM (6-carboxyfluorescein). The PCR reactions were conducted on Veriti 384 well PCR System (Applied Biosystems). PCR amplification was carried out in a total volume of 10 μL containing 2 × Taq PCR MasterMix (Gene Tech), 0.25 μM primer (each direction), and about 20 ng genomic DNA as the template. The genomic DNA of M. albus was extracted from tail muscle using the magnetic beads extraction Kit (NanoMagBio, China). DNA concentration was measured using a Nanodrop (ThermoFisher). A two-stage touchdown amplification program was used. An initial denaturation step of 5 min at 95°C was used, followed by a first stage in which the annealing temperature was gradually decreased from 62°C to 52°C through 10 cycles. This treatment was followed by a second stage consisting of 25 amplification cycles, in which the annealing temperature was set 52°C. Denaturation and elongation steps were held constant for 30 s at 95°C and 30 s at 72°C, respectively, during both stages. An elongation step of 20 min at 72°C was performed after stage 2. PCR products were diluted with ddH2O 10× (PCR product of multiplex mix I) and 20× (PCR product of multiplex mix II). Each diluted PCR product was then mixed with 8 μL of Hi-Di™ Formamide mixed with 0.5 μL of GeneScan 500 LIZ Size Standard (Thermo Fisher Scientific). Fragment lengths were determined using GeneMarker 2.2 (SoftGenetics, State College, USA) after capillary gel electrophoresing on an ABI 3730xl Genetic Analyzer (Life Technologies, Carlsbad, CA, USA).
The genotypic data were initially processed in Microsoft Office Excel 2007, manually checked for errors, and then transformed into input files that were required for subsequent analyses. The number of alleles (NA), the effective number of alleles (NE), observed heterozygosity (HO), expected heterozygosity (He), Shannon’s Information Index, and Fixation index (F) were calculated for each locus using GENALEX 6.5. 35 The polymorphism information content (PIC) was implemented by Excel-microsatellite-toolkit version 3.1. 36 Deviation from Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were tested using the exact tests of Genepop version 4.7.5 (https://kimura.univ-montp2.fr/∼rousset/Genepop.htm) with 10,000 dememorization and in 200 batches with 5000 iterations per batch. 37 The p values for HWE and LD were corrected for multiple comparisons by applying a sequential Bonferroni correction. MICROCHECKER v.2.2.3 38 was used to test for the possibility of scoring errors, allelic dropout, and null alleles.
Results
Identification of SSRs in the M. albus genome
The available 799.72 Mb genome sequences of M. albus were searched for the presence of different SSRs. A total of 364,802 SSRs were identified, and compound types (43,851) constituted 12.02% of the total SSRs. The total lengths of the identified SSRs were 8,204,641 bp, accounting for 1.03% of the whole genome length. The frequency and density of SSRs were 456 loci/Mb and 10,259.35 bp/Mb, respectively. Of 287,884 perfect SSRs (Table 1 and Figure 1), mononucleotide repeats were represented in maximum counts (121,595) followed by di- (93,972), tri- (19,499), tetra- (18,675), penta- (26,757), and hexa-nucleotides (7386).
The number, length, frequency, and density of six different types of perfect SSRs.
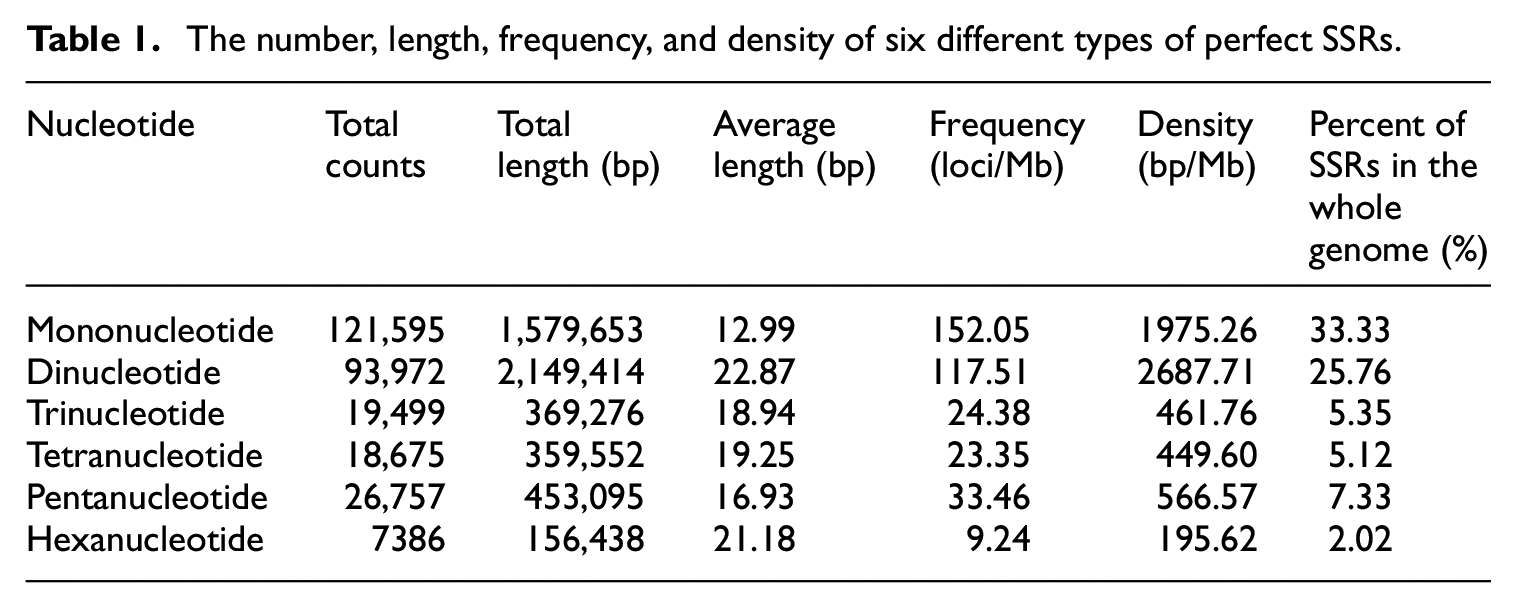

Distributions of SSRs in the whole genome of swamp eel (M. albus).
We detected 413 types of SSR motifs in total (Supplemental Table 1). There were 2, 4, 10, 32, 100, 265 types of mono- to hexanucleotide repeats in M. albus, respectively. Among different types of repeats, the motif A had the highest occurrence (37.63%) in the eel genome, followed by AC (24.31%), AG (4.87%), C (4.6%), AT (3.4%) (Supplemental Table 1). The most frequent mono- to hexa-nucleotides motifs were A (89.09%), AC (74.47%), AAT (37.65%), ATCC (24.61%), AAATC (25.57%), and ACACAG (22.22%) (Figure 2). Among dinucleotide repeats, the motif AC had the highest occurrence (74.47%), followed by AG (14.92%) and AT (10.53%), and CG repeats were the lowest in proportion (0.07%). Amongst the 10 types of trinucleotide repeats identified, AAT repeats were the most abundant (37.65%), followed by AGG (12.99%), AAG (11.44%), AAC (10.77%), AGC (9.82%), ATC (9.51%), ACC (7.83%), and the other three repeats (ACT, ACG, and CCG) were uncommon (together representing 3.91%). For tetra-, penta-, and hexanucleotide repeats, the most abundant motifs were ATCC (24.61%), AAATC (25.57%), and ACACAG (22.22%), respectively.
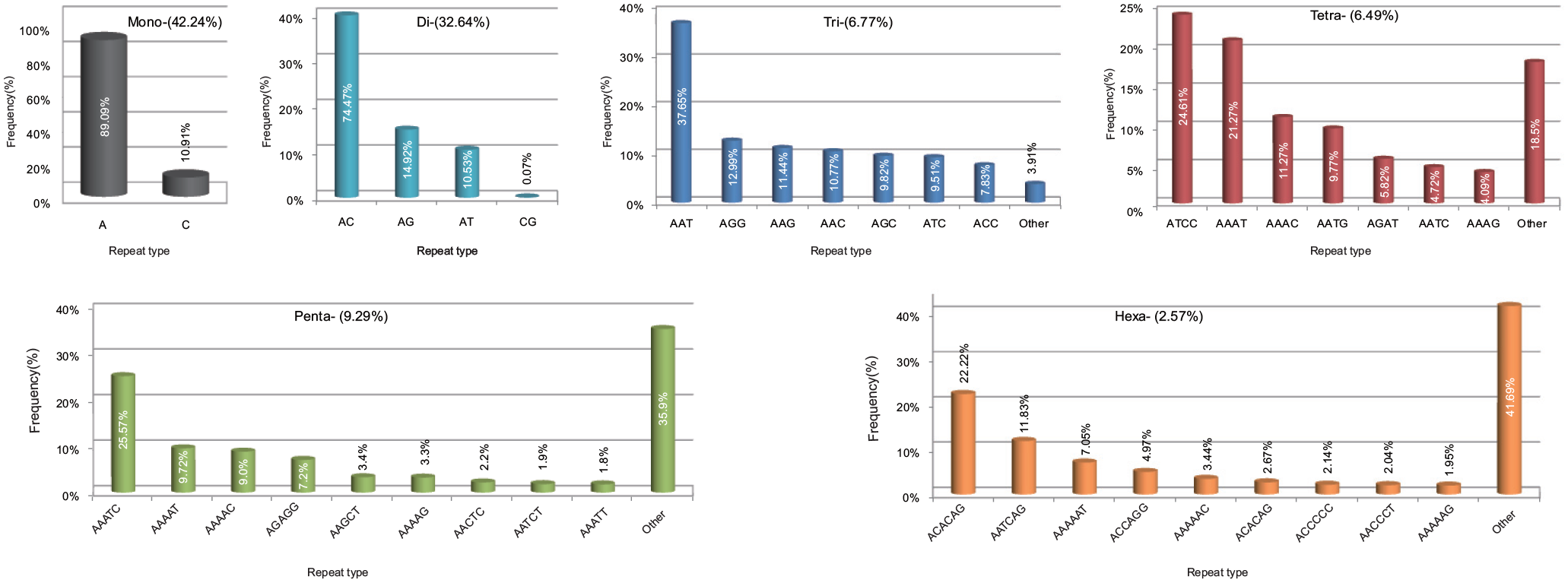
The distribution of the major repeat types in the genome of M. albus.
We then analyzed the distributions of SSRs on each chromosome of the swamp eel genome. A total of 316,657 SSR markers were distributed on 12 chromosomes and the number of SSRs on the chromosomes ranged from 18,962 to 35,822 (Table 2). The average frequency of SSRs was 404.32 SSRs per Mb, ranging from 381.49 SSR per Mb on chromosome 11 to 407.47 SSR per Mb on chromosome 8 (Table 2). Noticeably, when mononucleotide SSRs was not considered, the highest frequency of SSR markers was found on chromosome 3 and the least one was on Chromosome 12 (Table 2). Besides, about 21% of the identified SSRs were present in unanchored scaffolds and contigs (data not shown). The percentage of mononucleotide repeats was the highest, followed by di-, and pentanucleotide repeats. The percentage of tri- and tetranucleotides were nearly equivalent, and the percentage of hexanucleotide repeats was the lowest (Supplemental Figure 1).
Number of SSRs on the 12 chromosome of M. albus.
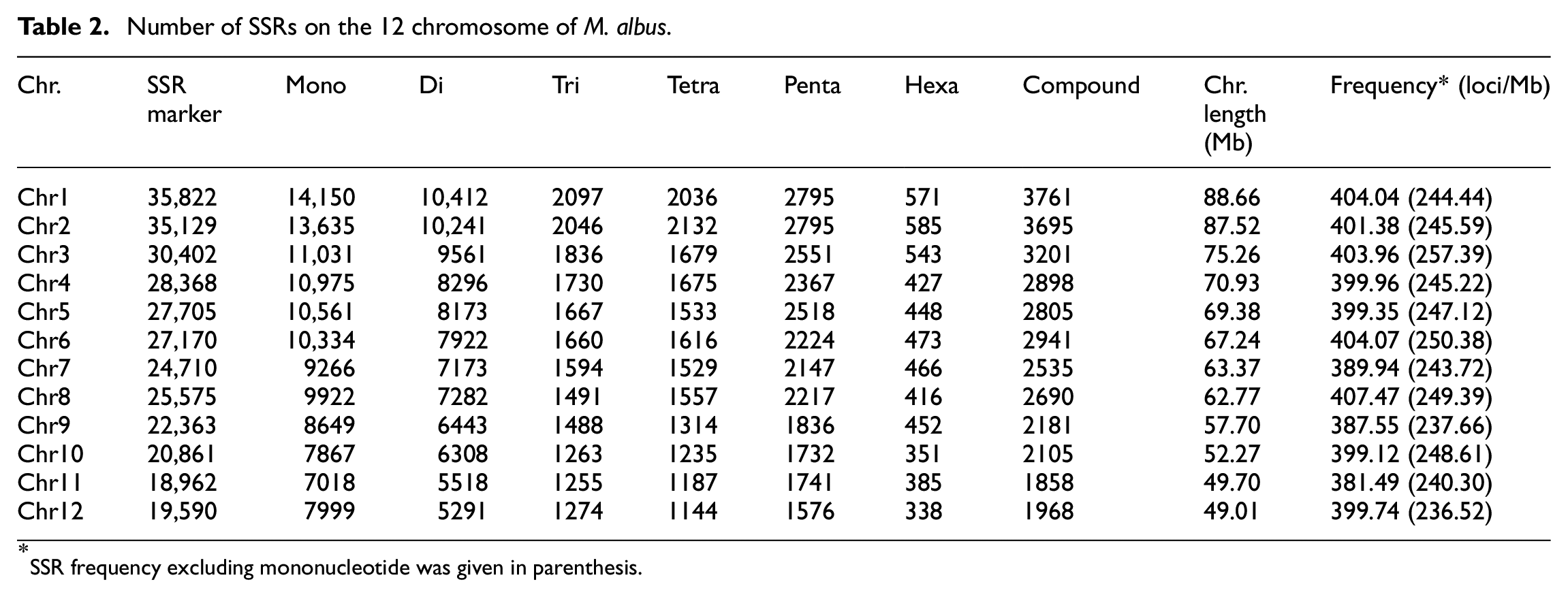
SSR frequency excluding mononucleotide was given in parenthesis.
The distribution of SSRs in different genomic regions, including exons, 5′UTR, 3′UTR, CDSs, intronic, and intergenic regions, were determined. The occurrences and relative frequencies of SSRs were found to differ significantly in coding and noncoding regions (Figure 3(a)). SSRs were most commonly located in intergenic regions, followed in order by intronic regions, exons, and CDSs (Figure 3(b)). Noticeably, the frequencies of SSRs in 3′UTRs (13.27 SSRs/Mb) are higher than those in 5′UTRs (2.52 SSRs/Mb) and CDSs (2.87 SSRs/Mb) (Figure 3(b)).

Comparisons of percentage (a) and frequency (b) of SSRs in different genomic regions of M. albus.
In CDSs, trinucleotide SSRs were the most abundant type, followed by hexa-, di-, penta-, mono-, and tetra- nucleotide SSRs (Figure 4(a)). In exons, mononucleotide SSRs were the most abundant type, followed by di-, tri-, penta-, tetra-, and hexa-nucleotide SSRs (Figure 4(b)). In intronic and intergenic regions, mononucleotide SSRs were the most abundant type, followed in decreasing order by di-, penta-, tetra-, and tri-nucleotide SSRs, and hexanucleotide SSRs were the least abundant type (Figure 4(c) and (d)).
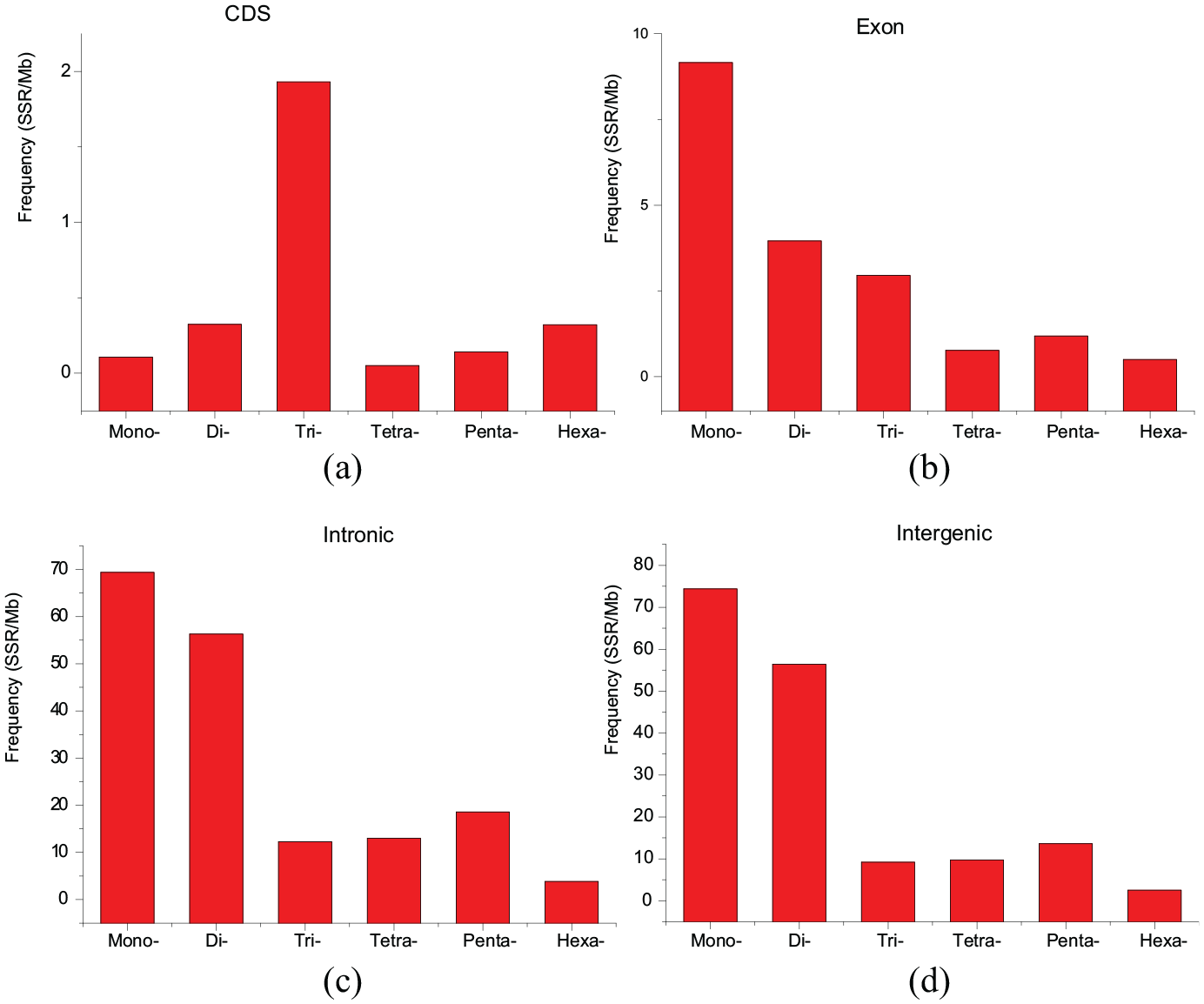
Relative frequency of mono- to hexa-nucleotide SSRs in different genomic regions of the swamp eel genomes. (a–d) Represent CDSs, exons, intronic regions, and intergenic regions, respectively.
Development of the genome-wide SSR primers
The flanking sequences of all identified SSRs were used to design suitable forward and reverse primer pairs. A total of 284,775 SSR markers were successfully developed on 12 chromosomes in swamp eel, which accounted for 78.02% of all identified SSRs. Based on the start positions of SSR markers, we successfully anchored these markers to the reference genome physical map, and there is no great difference of marker density among chromosomes on the physical map (Figure 5). This high-density physical map would provide an opportunity for accelerating mapping and breeding applications of different traits.

Overview of the high-density SSR physical map in swamp eel (M. albus). The bar represents the number of SSR markers within a 1-Mb window.
Screen and validation of polymorphic SSRs
The sequenced genome and reported transcriptome provided opportunities for identifying the repeat size variations rapidly. As genotyping mononucleotide SSRs can be error prone, we only selected from di- to hexanucleotide repeats (162,612 SSRs) to screen polymorphic SSRs by using PSR tool, and 871 SSRs showed polymorphisms across those 12 individuals among those perfect SSRs (Supplemental Table 2).
Among 101 SSR loci tested, 63 loci were excluded due to PCR failure, monomorphism, unexpected product size, or multiple peak profiles in the following genotyping analysis. The other 38 ones were validated to be polymorphic among six individuals and used for genotype analysis furtherly (Table 3).
Characteristics of 38 SSR loci developed for 29 samples of M. albus.

F: fixation index; (He-Ho)/He: 1 – (Ho/He); He: expected heterozygosity; Ho: observed heterozygosity; I: Shannon’s information index; N: number of samples; Na: number of different alleles; Ne: number of effective alleles; PIC: polymorphism information content; P of HW: Hardy-Weinberg probability test; uHe: unbiased expected heterozygosity.
Indicated deviation from Hardy-Weinberg equilibrium (p < 0.05) after Bonferroni’s correction.
Genetic variations at 38 loci were assessed in a panel of 29 individuals of our breeding population. The number of alleles per locus (NA), number of effective alleles per locus (NE), Shannon’s information index (I), observed (HO) and expected (He) heterozygosity, unbiased expected heterozygosity (uHe), fixation index (F), and polymorphic information content (PIC) are presented in Table 3. The PIC values ranged from 0.066 to 0.896 (0.507 in average). Twenty SSR markers were highly informative (PIC > 0.5), and 14 ones were in medium polymorphism degree (0.5 > PIC > 0.25). A total of 201 alleles were identified in 38 loci. NA varied from 2 to 13 (5.29 in average), and NE ranged from 1.072 to 10.447 (2.765 in average) per locus. The Shannon information index (I) ranged from 0.174 to 2.432, with a mean of 1.09. HO and HE ranged from 0.069 to 0.828 and from 0.067 to 0.904, respectively. The fixation index (F) ranged from −0.091 to 0.477 with an average of 0.152, and 19 loci presented heterozygote deficiencies (FIS > 0.15). Only two of the 38 loci (Ma30 and Ma50) showed significant deviations from HWE after sequential Holm-Bonferroni corrections (p < 0.05). Besides, only one significant deviate from linkage equilibrium (loci Ma65 and Ma71) (following the sequential Holm-Bonferroni correction, p < 0.05) of the 703 pairwise comparisons in the tested population, and none of the remaining markers showed significant LD (p > 0.06).
Discussion
In this study, a total of 364,802 SSRs were identified based on the complete genome of M. albus. Even only considering from di- to hexanucleotide repeats, the identified number (223,012) and frequency (278 loci/Mb) of SSRs in this study is much larger than the previous reports obtained by using restriction site associated DNA (RAD) sequencing (9897 loci, 195 loci/Mb) 25 or second generation genome sequencing (99,293 loci, 179 loci/Mb). 26 Considering the criteria selected for identifying the SSRs in our work are the same as those used previously, 26 such great number of SSRs identified here might be attribute to the high continuity of the assembled genome based on PacBio and Hi-C sequencing. The total length of all SSRs from M. albus genome was 8.2 Mb, and positive correlations between the genome size and total length of SSRs (R2 = 0.773, p < 0.01) were also confirmed when compared with those marine fish reported recently, 8 suggesting that this rule might also work in teleost fishes.
The motif A was the most abundant mononucleotide motif in swamp eel, which is also found in different mosquitoes, 39 and some marine animals. 11 The most abundant repeat motifs of di- and trinucleotide repeats seem to be conserved. AC was the most abundant repeat categories in dinucleotides and CG was the least one, which are consistent with the cases in most vertebrates reported by Jiang et al. 11 and in other eukaryotes. 40 AAT was the predominant trinucleotides type, which is similar to the situation in human, 41 camelid, 42 and many teleost fishes.8,11 Interestingly, the higher proportions of penta-nucleotide than tri- and tetranucleotide motifs are consistent with the result characterized by using RAD sequencing, 25 but different from those reported by Li et al., 26 and the proportions of each repeat motifs of tri-, tetra-, penta-, and hexanucleotide repeats are different from those reported in two recent works about swamp eel.25,26 Whether such discrepancy could be explained by using different sets of germplasm needed to be evaluated in future.
SSRs are powerful markers for population genetic analysis, QTL mapping, and other related genetic and genomic studies. 43 Here, 284,775 SSR markers were developed successfully, and a highly density physical map was constructed, which might be used for future marker assisted breeding and QTL mapping. A total of 871 polymorphic SSRs were also obtained through comparing sequence variants among 12 public available transcriptomes, and 38 loci were validated by capillary gel electrophoresing. As SSRs with PIC more than 0.5 and He more than 0.6 are the most reasonable informative loci for application in genetics, 44 20 SSRs are highly informative and would be very useful genetic markers for future genetic diversity, population genetic studies, and molecular breeding programs. Altogether, there was a high level of genetic diversity (Na = 5.3, Ho = 0.469, He = 0.554, PIC = 0.508) for the population studied here, which is similar to that of one wild population found in Jiangxi province (Na = 5.7, Ho = 0.3795, He = 0.5397, PIC = 0.4987), 25 but relatively lower than four wild populations found in Hubei, Anhui, Jiangsu, and Zhejiang provinces along the middle and lower reaches of the Yangtze River Basin. 45 Considering the SSRs developed in this study were mainly trinucleotide repeats, which are typically associated with a low level of variability,46,47 the allele number (NA) and PIC (Table 3) suggest that the genetic diversity within this broodstock is not low in contrast to those mainly using dinucleotide repeats. 45 Noticeably, the average Ho (0.467) was slightly less than the He (0.553), and the mean value of the fixation index (F) was 0.152, indicating that a deficiency of heterozygotes and inbreeding within this breeding population.
Conclusion
We identified 364,802 SSRs which belong to 413 types of motifs, and developed 284,775 potential SSR markers accordingly. A total of 871 polymorphic SSRs were identified and 38 ones were validated. These newly identified SSRs and developed high variable SSR markers would enrich SSR marker resources and provide a resourceful dataset for genetic, genomic, and evolutionary biology studies of this species along with SSR markers from previous reports. Using 38 validated polymorphic SSR loci, the high level of genetic diversity and existence of inbreeding were revealed in our breeding population, which would provide a background of knowledge for future breeding programs.
Supplemental Material
sj-pdf-1-sci-10.1177_00368504211035597 – Supplemental material for Genome-wide identification of simple sequence repeats and development of polymorphic SSR markers in swamp eel (Monopterus albus)
Supplemental material, sj-pdf-1-sci-10.1177_00368504211035597 for Genome-wide identification of simple sequence repeats and development of polymorphic SSR markers in swamp eel (Monopterus albus) by Hai-feng Tian, Qiao-mu Hu and Zhong Li in Science Progress
Supplemental Material
sj-pdf-2-sci-10.1177_00368504211035597 – Supplemental material for Genome-wide identification of simple sequence repeats and development of polymorphic SSR markers in swamp eel (Monopterus albus)
Supplemental material, sj-pdf-2-sci-10.1177_00368504211035597 for Genome-wide identification of simple sequence repeats and development of polymorphic SSR markers in swamp eel (Monopterus albus) by Hai-feng Tian, Qiao-mu Hu and Zhong Li in Science Progress
Supplemental Material
sj-pdf-3-sci-10.1177_00368504211035597 – Supplemental material for Genome-wide identification of simple sequence repeats and development of polymorphic SSR markers in swamp eel (Monopterus albus)
Supplemental material, sj-pdf-3-sci-10.1177_00368504211035597 for Genome-wide identification of simple sequence repeats and development of polymorphic SSR markers in swamp eel (Monopterus albus) by Hai-feng Tian, Qiao-mu Hu and Zhong Li in Science Progress
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Central Public-Interest Scientific Institution Basal Research Fund, Chinese Academy of Fishery Sciences (CASF) (Grant numbers 2020XT08 and 2020TD33).
Supplemental material
Supplemental material for this article is available online.
Author biographies
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.