
Abstract
Acute lung injury (ALI) is one of the most prevalent respiratory syndromes of excessive inflammatory reaction during lung infection. Candida albicans (C. albicans) infection is among the leading causes of ALI. MicroRNAs (miRNAs) regulate the expression of target mRNAs, including those involved in inflammatory processes, by binding to the 3′UTR. To date, the roles of miRNAs in C. albicans-induced ALI remain unclear. In this study, we investigated the role of miR-384-5p in C. albicans-induced ALI and its underlying molecular mechanism. RT-PCR, Western blot, ELISA, Myeloperoxidase (MPO) assay, microRNA target analysis, transient transfection, and luciferase reporter assay were utilized. In vivo study was conducted on mouse model. The expression of miR-384-5p was upregulated and positively correlated with inflammatory cytokine production in lung tissues and RAW264.7 and J774A.1 macrophages infected with C. albicans. The miR-384-5p inhibitor alleviated the inflammatory reaction induced by C. albicans. Target prediction analysis revealed that PGC1β was a target of miR-384-5p, which was further validated by the PGC1β 3′-UTR luciferase assay and the inverse correlation between the expression of miR-384-5p and PGC1β in C. albicans-infected ALI tissues and macrophages. Moreover, macrophages transfected with miR-384-5p mimic exhibited reduced levels of PGC1β. The suppression of the expression of PGC1β by C. albicans infection in the macrophages was abrogated by miR-384-5p inhibitor. Then, we demonstrated that PGC1β played an inhibitory role in C. albicans-induced production of inflammatory cytokines. Furthermore, suppression of miR-384-5p in macrophages inhibited the activation of the NF-κB, MAPK, and Akt signaling pathways triggered by C. albicans, but not the STAT3 pathway. These results demonstrate that miR-384-5p contributes to C. albicans-induced ALI at least in part by targeting PGC1β and enhancing the activation of the NF-κB, MAPK, and Akt inflammatory signaling pathways. Thus, targeting miR-384-5p might exert a protective effect on C. albicans-induced ALI.
Introduction
C. albicans is an opportunistic pathogen in humans and other mammals and is the most abundant species in the Candida genus. C. albicans infection contributes to 80% of nosocomial infections caused by fungal pathogens. 1 C. albicans infection is highly prevalent and causes high mortality in immunocompromised individuals.2–6 C. albicans infection is an important cause of acute lung injury (ALI). Studies showed that C. albicans infection could enhance the production of inflammatory cytokines that play a pivotal role in ALI.7,8 Macrophages are critical cells in the pathogenesis of ALI that regulate inflammation by secreting inflammatory cytokines.9–12 However, the molecular mechanisms underlying the pathogenesis of C. albicans -induced ALI remain to be elucidated.
miR-384-5p was found to play an essential role in osteoarthritis by targeting SOX and negatively regulating age-related osteogenic differentiation of bone marrow mesenchymal cells.13,14 miR-384-5p is also required for spine plasticity and long-term potentiation. 15 Jiang et al. 16 reported that miR-384-5p is involved in rotenone-induced neurotoxicity. Moreover, miR-384-5p regulates macrophage autophagy by controlling Beclin-1. 17 Also, miR-384-5p is a critical factor in cardioprotection in myocardial ischemia. 18 Fan et al. 19 showed that miR-384-5p enhanced the function of endothelial progenitor cells (EPCs) in cerebral ischemic stroke by targeting by small interference RNA to Delta-like ligand 4 (si-DLL4). Moreover, enhanced macrophage autophagy can be induced by downregulating miR-384-5p in inflammatory macrophages, contributing to the development of diabetic encephalopathy. 20 However, the roles of miR-384-5p in inflammation and C. albicans-induced ALI remain unclear.
The PPARγ-coactivator-1 (PGC1) family was initially identified as a significant regulator of oxidative metabolism.21,22 Lin et al. 23 showed that the PGC1 family comprises transcriptional coactivators that are highly versatile in their functions and connect lipid homeostasis with inflammation. Being a member in the PGC1 family, PGC1β has been identified as one of the inducible molecules for inhibiting inflammation in gout.24,25 As reported by Vats et al., 26 transgenic overexpression of PGC1β strongly inhibited the proinflammatory reaction in LPS-challenged macrophages, whereas knockdown of PGC1β reversed the inhibition.
Herein we investigated the potential role of miR-384-5p in C. albicans-induced ALI. We found that miR-384-5p was involved in C. albicans-induced ALI at least in part by suppressing the expression of PGC1β and contributed to the activation of TLR4-mediated inflammatory signaling pathways triggered by C. albicans infection. These findings reveal a new mechanism by which the miR-384-5p-PGC1β pathway regulates C. albicans-induced inflammation in ALI, which may provide evidence for developing new approaches for the treatment of C. albicans-induced ALI.
Materials and methods
Animal experiments
Animal experiments was conducted according to the guidelines provided by the experimental animal center of Wenzhou Medical University, Wenzhou, China, and approved by the Animal Care and Use Committee of Wenzhou Medical University, Wenzhou, China. BALB/c mice (Male, 8–10 weeks old) were housed five per cage at 22–25°C on a 12-h light/dark cycle. A total of 56 mice were divided into seven groups (eight mice per group): No treatment, PBS control, C. albicans, PBS+miR-384-5p inhibitor scrambled control, PBS+miR-384-5p inhibitor (Sigma Aldrich, USA; catalog #: MSTUD0435), C. albicans+ miR-384-5p inhibitor scrambled control, and C. albicans+miR-384-5p inhibitor. PBS, C. albicans (1 × 107 CFU in 60 μl of PBS), solution of miR-384-5p inhibitor and scrambled control (5 μg/μl in 50 μl volume) were intratracheally injected into the mice at the same time. All mice were euthanized with CO2 24 h post C. albicans exposure for further experiments. The left lung was lavaged with the bronchoalveolar lavage fluid (BALF) collected.
Quantification of pulmonary edema
Wet/dry (W/D) weight ratio was determined to evaluate lung edema caused by C. albicans infection. 27 24 h after the mice were exposed to C. albicans, the mice were sacrificed, and the lungs were removed, weighted, and dried at 80°C for 24 h. The W/D ratio was then calculated.
Myeloperoxidase assay
Lung Myeloperoxidase (MPO) activity was determined to assess the infiltration of neutrophils in the lung tissues using a MPO colorimetric assay kit (Sigma-Aldrich; Catalog #: MAK068) according to the manufacture’s instructions. Whole lungs were homogenized in 2 ml of 50 mM potassium phosphate (KH2PO4) supplemented with 5 mM of EDTA and 5% hexadecyltrimethylammonium bromide (Sigma-Aldrich; Catalog#: H6269), sonicated and centrifuged at 12,000 × g for 20 min. The supernatant was collected and mixed with assay buffer (1:15). Values of Optical Density (OD) at 490 nm were read in a BioTek ELx808 microplate reader.
Cell culture
Mouse RAW264.7 cells and J774A.1 cells (ATCC, USA; Catalog#: TIB-71 and TIB-67) was cultured in high glucose Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen, USA; Catalog #: 220511) with 10% FBS (Gibco, USA; Catalog #: A31605), 100× penicillin-streptomycin (Thermo Fisher Scientific, USA; Catalog #: 10378016) at 37°C with 5% CO2. Before stimulating with C. albicans, the culture medium was replaced with fresh medium and 1 × 105 CFU of C. albicans suspended in DMEM was administered. The cells were cultured in the presence of C. albicans for 0, 1, 2, 6, 12, and 24 h. For co-treatment with C. albicans and miR-384-5p inhibitor, the RAW264.7 cells, and J774A.1 cells were treated with C. albicans for 24 h.
Extraction of RNA and real-time RT-PCR
Total RNA was extracted from lung tissues and the macrophages cell lines by using a mirVana isolation kit (Ambion, USA; Catalog #: AM1560). The levels of miR-384-5p were examined by qRT-PCR according to the TaqMan® MicroRNA Assay protocol (Applied Biosystems, USA; Catalog #: 4427975), and U6 snRNA (Applied Biosystems; Catalog#: 001973) served as an internal control. To examining the mRNA levels of IL-6, IL-1β, TNF-α, and TGF-β, the total RNA was reversely transcribed into equal amounts of cDNA using a SuperScript IV first-stand Synthesis kit (Thermo Fisher Scientific; Catalog #: 18091050) according to the manufacturer’s instructions. Real-time PCR was then performed in triplicates using an Applied Biosystems PowerUp SYBR Green Master Mix PCR kit (Applied Biosystems, USA; Catalog #: A25741) in an ABI 7300 real-time PCR system. The specific primers for miR-384-5p, IL-6, IL-1β, TNF-α, and TGF-β13,15,28 are shown in Table 1. GAPDH served as an internal control for IL-6, IL-1β, TNF-α, and TGF- β. The PCR data were analyzed with the 2-ΔΔCt method. 29
Primers for RT-PCR.

ELISA
Lung tissues and macrophage cells were lysed with lysis buffer (Thermo Fisher Scientific; Catalog #: FNN0011) and centrifuged at 12,000 rpm for 15 min at 4°C. The supernatant was then collected for quantifying IL-6, IL-1β, TNF-α, and TGF β levels using ELISA kits (IL-6 and TNF-α ELISA kits from BD Biosciences, USA; Catolog #: 550950, 554416; TGF-β and IL-1β ELISA kits from Thermo Fisher Scientific, Catalog #: BMS608-4TWO, BMS6002) according to the manufacture’s instructions. OD of each well was determined at 450 nm using a BioTek ELx808 microplate reader.
Transfection
RAW264.7 and J774A.1 cells were cultured and reached 70% confluence in a six-well culture plate before transfection. miR-384-5p inhibitor, inhibitor control, miR-384-5p mimic, and its scrambled control (Thermo Fisher Scientific; Catalog #: 4464066, 4464058) or pcDNA-PGC1β (Addgene, USA; Catalog #: 103125) were transfected into cells using Lipofectamine LTX reagent (Thermo Fisher Scientific; Catalog #: A12621) according to the manufacturer’s instructions. After 6 h transfection, the culture medium was replaced with fresh complete medium and cultured for additional 48 h. The cells and culture supernatants were collected for subsequent assays.
Luciferase reporter assay
To construct a wild type PGC1β 3-UTR luciferase reporter vector, the fragment of 3′UTR of PGC1β mRNA containing the binding site for miR-384-5p or mutant type of PGC1β 3′-UTR was cloned into pGL3 plasmid (The plasmids were from and stored in our department). The vectors were then co-transfected with miR-384-5p mimic into the macrophages using Lipofectamine LTX in a 96-well culture plate. 48 h post transfection, the cells were lysed, and luciferase activity was assessed using a dual-luciferase reporter assay system (Promega, USA; Catalog #: E1910).
Western blot analysis
Total proteins were extracted from lung tissues and macrophages with treatments using lysis buffer (Thermo Fisher Scientific) according to the manufacturers’ instructions. The concentrations of protein samples were measured with a BCA assay kit (Thermo Fisher Scientific; Catalog #: 23225). Total 50 μg of protein samples were loaded, separated in 10% SDS-PAGE gels, and transferred onto PVDF membranes (EMD Millipore, USA; Catalog #: IPFL85R). The membranes were blocked with 5% nonfat milk or 3% BSA in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, and 0.5% Tween 20) for 1 h. After washing with TBST, the membranes were incubated with the primary antibodies overnight at 4°C: PGC1β (1:1000) (Abcam, USA; Catalog #: AB176328), NF-κB (Catalog #: 4764), phospho-NF-κB (Ser536) (Catalog #: 3033), mitogen-activated protein kinase (MAPK) (Catalog #: 9212), phospho-MAPK (Thr202/Tyr204) (Catalog #: 98168), Akt (Catalog #: 9272), phospho-Akt (Thr308) (Catalog #: 13038), signal transducer and activator transcription 3 (STAT3) (Catalog #: 12640), phospho-STAT3 (Tyr705) (1:1000) (Catalog #: 9145) (Cell Signaling, USA), and GAPDH (1:40000) (Sigma-Aldrich) (Catalog #: G8795). The membranes were then washed and incubated with HRP-conjugated secondary antibodies (1:5000) (EMD Millipore) (Catalog #: AP307P) for 1 h at room temperature. Blots were visualized using Chemiluminescent substrates (Thermo Fisher Scientific) (Catalog #: 34577), and densitometry of the bands was analyzed using Image J software.
Statistical analysis
Quantified data are presented as mean ± S.D. Statistical significance was determined by unpaired Student’s two-tailed t-test or one-way ANOVA analysis with Bonferrorni post-hoc test, with p < 0.05 was considered to be statistically significant. The experiments were repeated at least three times.
Results
C. albicans infection upregulates the expression of miR-384-5p in mouse ALI tissues and macrophages
As shown in Figure 1(a), C. albicans induced an increase in the W/D ratio of lung weight 24 h after infection. A dramatically raised number of macrophages was detected in BALF (Figure 1(b)). MPO activity was also increased in infected mice (Figure 1(c)). The mRNA levels and secretion of proinflammatory cytokines IL-6, IL-1β, TNF-α, and TGF-β, a critical mediator of acute lung injury, 30 were significantly higher in ALI tissues than in the PBS control group (Figure 1(d)). As shown in Figure 1(e), the levels of miR-384-5p were dramatically upregulated in C. albicans-stimulated lung tissues (Figure 1(e)). To further examine the expression of miR-384-5p in vitro, we explored the expression of miR-384-5p at different time points (0 h, 1 h, 2 h, 6 h, 12 h, and 24 h after C. albicans treatment in RAW264.7 and J774A.1 macrophages). A time-dependent upregulation of miR-384-5p in response to C. albicans stimulation was observed in both RAW 264.7 and J774.1 cells (Figure 1(e)).

Upregulation of miR-384-5p induced by C. albicans in mouse ALI tissues and macrophages. (a) W/D ratios of lungs from mice infected with C. albicans (n = 6). (b) Amounts of macrophages in BALF were calculated (n = 6). (c) MPO activity to determine the infiltration of neutrophils in the lung tissues (n = 6). (d) Analysis of the levels of IL-6, IL-1β, TNF-α, and TGF-β in ALI tissues from mice infected with C. albicans by RT-PCR and ELISA. GAPDH was an internal control for RT-PCR. (e) Expression of miR-384-5p in ALI tissues and the RAW 264.7 and J774A.1 macrophages infected with C. albicans. The levels of miR-384-5p were assessed in ALI tissues harvested 24 h after C. albicans administration and in macrophages treated with C. albicans for 1, 2, 6, 12, and 24 h. The levels of miR-384-5p were normalized to U6 snRNA levels. Data are expressed as the mean ± S.D. of three independent experiments. Statistical analysis was performed using unpaired student’s two-tailed t-test for (a), (b), (c), (d) and expression of miR-384-5p in ALI tissues in (e), and one-way ANOVA analysis with Bonferrorni post-hoc test for expression of miR-384-5p in the RAW 264.7 and J774A.1 macrophages infected with C. albicans in E.
miR-384-5p promotes C. albicans-induced inflammation by enhancing the production of inflammatory cytokines in vitro and in vivo
To further confirm the roles of miR-384-5p in inflammation induced by C. albicans, a miR-384-5p inhibitor was transfected to suppress the expression of miR-384-5p in macrophages stimulated with C. albicans. As shown in Figure 2(a), the upregulation of miR-384-5p induced by C. albicans stimulation was significantly suppressed by the miR-384-5p inhibitor. Targeting miR-384-5p reduced the production of IL-6, IL-1β, TNF-α, and TGF-β by more than 2-fold compared to that in the scrambled control (Figure 2(b)). We then further investigated the effect of targeting miR-384-5p in mice infected with C. albicans. We found that the levels of IL-6, IL-1β, TNF-α, and TGF-β were significantly lower in the group treated with C. albicans and miR-384-5p inhibitor than in the group treated with C. albicans and scrambled control (Figure 2(c)). These findings suggest that targeting miR-384-5p can abrogate the inflammatory response triggered by C. albicans. Moreover, a miR-384-5p mimic was utilized to overexpress miR-384-5p in macrophages without C. albicans infection (Figure 2(d)). As expected, the miR-384-5p mimic enhanced the production of inflammatory cytokines (Figure 2(e)), indicating that miR-384-5p can promote C. albicans-induced inflammation by improving the production of inflammatory cytokines.

Involvement of miR-384-5p in C. albicans-induced inflammation in vitro and in vivo. (a) Changes in miR-384-5p levels in the RAW264.7 and J774A.1 macrophages transfected with miR-384-5p inhibitor followed by C. albicans treatment. The upregulation of miR-384-5p induced by C. albicans was effectively suppressed by the miR-384-5p inhibitor. (b) Changes in the production of IL-6, IL-1α, TNF-β, and TGF-α in the C. albicans-infected RAW264.7 and J774A.1 macrophages transfected with the miR-384-5p inhibitor. (c) Production of IL-6, IL-1α, TNF-β, and TGF-α in lung tissues from mice administered with C. albicans and miR-384-5p inhibitor. (d) The overexpression of miR-384-5p in the RAW264.7 and J774A.1 macrophages by transfection of its mimic was confirmed by RT-PCR. (e) Changes in the production of IL-6, IL-1α, TNF-β, and TGF-α in the RAW264.7 and J774A.1 macrophages transfected with the miR-384-5p mimic. Data are expressed as the mean ± S.D. of three independent experiments. Statistical analysis was performed using one-way ANOVA analysis with Bonferrorni post-hoc test.
PGC1β is a target of miR-384-5p
We then sought to explore the molecular mechanism underlying the stimulatory effect of miR-384-5p on C. albicans-induced inflammation. PGC1β was predicted as a target of miR-384-5p in microRNA.org, TargetScanMouse and miRDB. The prediction analysis confirmed the presence of specific binding sequences of miR-384-5p in the 3′-UTR of PGC1β (Figure 3(a)), which was further validated by a luciferase reporter assay. Macrophages were co-transfected with miR-384-5p mimic and wild-type pGL3-PGC1β-3′-UTR (WT) or mutated pGL-3-PGC1β-3′-UTR (Mut). The miR-384-5p mimic remarkably suppressed the luciferase activity of the WT 3′-UTR reporter but not the mutant 3′-UTR reporter (Figure 3(b)). As shown in Figures 3(c) and 1(e), the expression of PGC1β was inversely correlated with the levels of miR-384-5p in C. albicans-induced ALI tissue and macrophages stimulated with C. albicans. Moreover, macrophages transfected with the miR-384-5p mimic exhibited reduced levels of PGC1β (Figure 3(d)). Furthermore, the rescue of PGC1β levels was observed in RAW264.7 and J774A.1 macrophages co-treated with C. albicans and miR-384-5p inhibitor (Figure 3(e)). These findings indicate that miR-384-5p can downregulate the expression of PGC1β at the post-transcriptional level by binding to its 3′-UTR.

PGC1β is a target gene of miR-384-5p. (A) The binding site of miR-384-5p in the PGC1β 3′-UTR predicted using a microRNA program. (B) The RAW264.7 and J774A.1 macrophages were co-transfected with PGC1β WT 3′-UTR or mutant 3′-UTR luciferase reporter vector and miR-384-5p mimic or control mimic. The luciferase activity was measured and normalized to renilla luciferase activity in the assay. (C) Western blot of PGC1β in ALI tissues induced by C. albicans and macrophages infected with C. albicans for 1, 2, 6, 12 and 24 h. PGC1β was downregulated in ALI tissues, and RAW264.7 and J774A.1 exhibited a time-dependent reduction in the levels of PGC1β in response to C. albicans treatment. (d) The expression of PGC1β in the RAW264.7 and J774A.1 macrophages transfected with miR-384-5p mimic. (e) The expression of PGC1β in the RAW264.7 and J774A.1 macrophages treated with C. albicans and transfected with miR-384-5p inhibitor. Densitometry analysis of the Western blot represents the ratio of PGC1β to GAPDH. Data are expressed as the mean ± S.D. of three independent experiments. Statistical analysis was performed using one-way ANOVA analysis with Bonferrorni post-hoc test.
PGC1β negatively regulates C. albicans-induced inflammation
PGC1β was identified as one of the negative regulators in the proinflammatory reaction, NLRP3 inflammasome activation, and development of inflammatory phenotype.24–26 To confirm that the stimulatory effect of miR-384-5p is mediated by targeting PGC1β, PGC1β was overexpressed by transfection of macrophages with pcDNA-PGC1β. The overexpression of PGC1β was confirmed by Western blot (Figure 4(a)). As shown in Figure 4(b), the enhanced production of IL-6, IL-1β, TNF-α, and TGF-β triggered by C. albicans was abrogated by the forced overexpression of PGC1β, which was consistent with the results in macrophages with miR-384-5p knockdown (Figure 2(b)). These results indicate that PGC1β plays an inhibitory role in modulating the stimulatory effect of C. albicans on inflammation. Thus, targeting PGC1β could promote C. albicans-induced inflammation.

Transgenic overexpression of PGC1β suppressed inflammatory responses to C. albicans in macrophages. (a) Transgenic overexpression of PGC1β in RAW264.7 cells was examined by Western blot. The densitometry analysis of the Western blot results represents the ratio of PGC1β to GAPDH. (b) Changes in the production of IL-6, IL-1β, TNF-α and TGF-β in C. albicans-infected RAW264.7 cells with transgenic PGC1β over-expression. Data are expressed as the mean ± S.D. of three independent experiments. Statistical analysis was performed using one-way ANOVA analysis with Bonferrorni post-hoc test.
Suppression of miR-384-5p inhibits the activation of the NF-κB, MAPK, and Akt signaling pathways triggered by C. albicans but not the STAT3 pathway
It has been reported that C. albicans can activate the pulmonary NF-κB, MAPK, Akt, and STAT3 signaling pathways to regulate inflammation in normal and immunosuppressed mice. 31 To further investigate the mechanism underlying the stimulatory role of miR-384-5p in the C. albicans-induced inflammatory response, the levels of phospho-NF-κB, phospho-MAPK, phospho-Akt and phospho-STAT3 were examined in RAW264.7 cells infected with C. albicans and transfected with the miR-384-5p inhibitor. As shown in Figure 5, suppression of miR-384-5p by its inhibitor significantly abolished the stimulatory effect of C. albicans on activation of the NF-κB, MAPK, and Akt signaling pathways but not the STAT3 pathway. Thus, we speculated that miR-384-5p might be implicated in several C. albicans-triggered signaling pathways to modulate the development of inflammation.
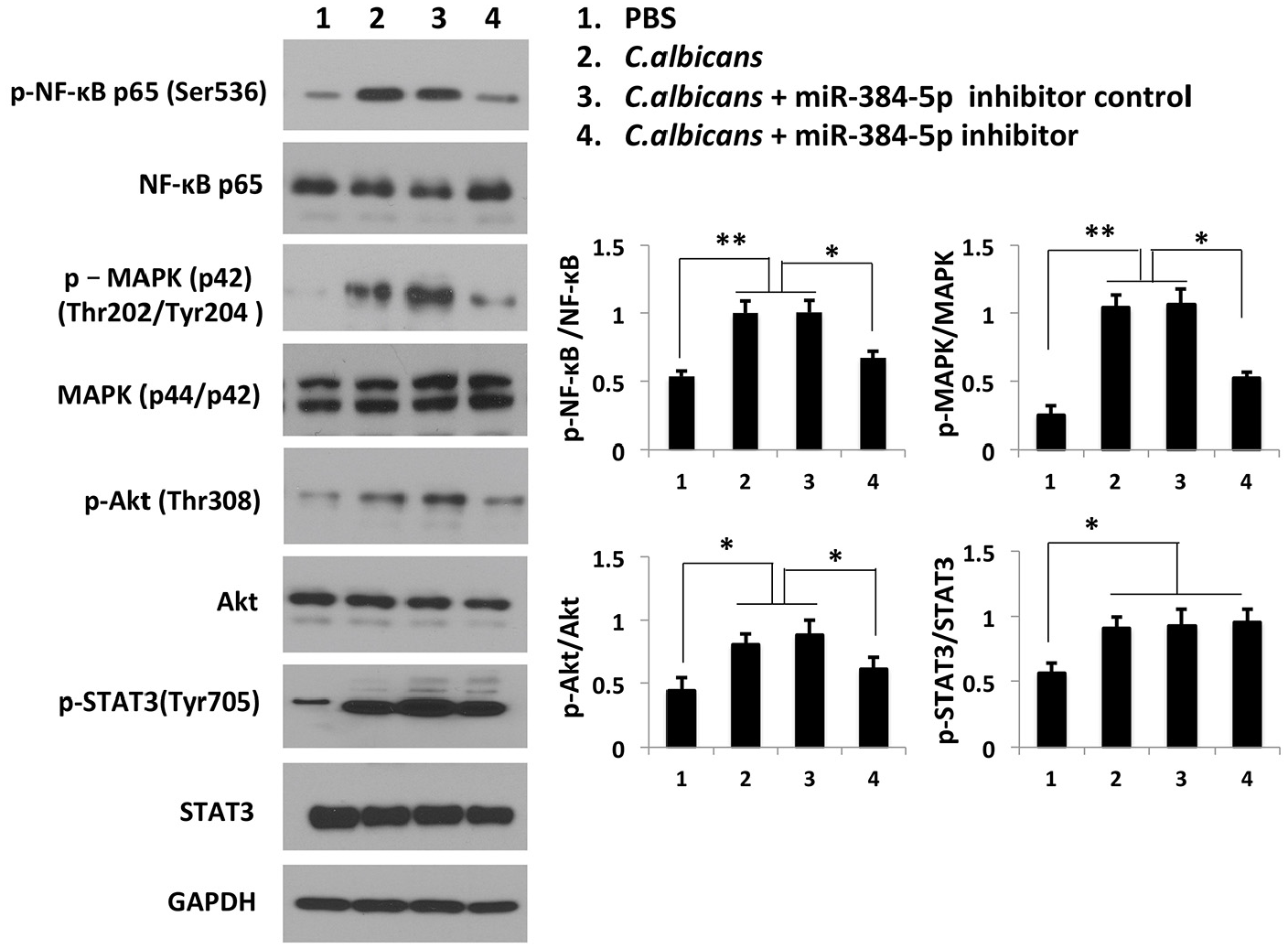
Targeting miR-384-5p inhibits the activation of inflammatory signaling pathways triggered by C. albicans. C. albicans activated the NF-κB, MAPK, Akt, and STAT3 pathways in RAW264.7 cells, as evidenced by the increases in phosphorylated NF-κB, MAPK, Akt, and STAT3. Transfection with the miR-384-5p inhibitor blocked the phosphorylation of NF-κB, MAPK, and Akt but not STAT3. The densitometry analysis of the Western blot results represents the ratio of phospho-NF-κB, MAPK, Akt, or STAT3 to NF-κB, MAPK, Akt, or STAT3, respectively. Statistical analysis was performed using one-way ANOVA analysis with Bonferrorni post-hoc test.
Discussion
ALI is a common pulmonary inflammatory syndrome with a high mortality rate. C. albicans infection is among the leading causes of ALI. MiRNAs have been found to modulate inflammatory responses both negatively and positively in ALI.28,32,33 miR-384-5p has emerged as a new therapeutic target in neuronal damage and neurotoxicity,16,34 age-related bone loss, 14 and diabetic encephalopathy. 17 In this study, we evaluated the function of miR-384-5p in C. albicans-induced ALI and investigated the underlying molecular mechanism. Our findings indicated that miR-384-5p is involved in C. albicans-induced ALI by targeting PGC1β and participating in the activation of inflammatory signaling pathways in response to C. albicans. This study provides new insight into the development of novel therapeutic targets in C. albicans-induced pulmonary inflammatory diseases.
Systemic and excessive local inflammation is a major cause of ALI, during which the secretion of pro-inflammatory cytokines is remarkably increased in pulmonary tissues.31,35 TGF-β is a critical mediator of acute lung injury in the early inflammatory phase of ALI. 30 As reported by Sanjabi et al. 36 TGFβ played anti-inflammatory and proinflammatory roles in immunity and autoimmunity. TGF-β was reported as pulmonary pro-inflammatory cytokine in the study on signal pathways in C. albicans induced acute lung injury. 31 Han et al. 37 found that TGFβ mediated skin inflammation as a pro-inflammatory cytokine. Sureshbabu et al. 38 revealed that TGFβ promoted pulmonary inflammation. We found that the expression levels of TGF-β were upregulated at the early phase of C. albicans infection, and targeting miR-384-5p specifically inhibited the production of IL-6, IL-1β, TNF-α, and TGF-β. In our preliminary study, miR-384-5p inhibitor did not reduce the production of anti-inflammatory cytokines such as IL-4, IL-10, and IL-11. Thus, we deduce that TGFβ may play a proinflammatory role in the early inflammatory phase in C. albicans-induced acute lung injury as our data were collected within 24 h post C. albicans infection. TGF-β may act dominantly as an anti-inflammatory cytokine during the late fibroproliferative tissue repair phase.39,40
As expected, the mRNA levels of IL-1β, IL-6, TNF-α, and TGF-β were increased in response to C. albicans infection, and the protein levels were elevated as well. miR-384-5p was upregulated in C. albicans-infected lung tissues and macrophage cell lines treated with C. albicans. The increase in the levels of miR-384-5p was time-dependent, indicating that miR-384-5p was positively correlated with C. albicans infection. Targeting upregulated miR-384-5p by its inhibitor could abrogate the effect of C. albicans on inflammation, suggesting that miR-384-5p was implicated in the immune response triggered by C. albicans. It is worth noting that despite the low basal levels of miR-384-5p in macrophages and lung tissues, its inhibitor could not significantly suppress the basal production of IL-1β, IL-6, TNF-α, or TGF-β. Therefore, we deduced that the expression level of miR-384-5p was closely related to its regulatory effect on the inflammatory responses stimulated by C. albicans. In contrast, the low basal levels of miR-384-5p did not influence the production of cytokines. The miR-384-5p inhibitor did not completely abolish the effect of C. albicans in vivo and in vitro, which can be explained by the knockdown efficiency of the inhibitor and the existence of other mechanisms regulating inflammation induced by C. albicans infection.
Given the positive correlation between miR-384-5p and C. albicans infection, the effect of direct overexpression of miR-384-5p in macrophages was then investigated in vitro. We found that overexpression of miR-384-5p by transfection of its mimic could imitate C. albicans stimulation. miRNAs regulate their target genes at the post-transcriptional level by inducing mRNA degradation or blocking translation, which is the major regulatory mechanism of the specific expression of miRNAs. 41 PGC1β was predicted to be a target of miR-384-5p. When macrophages were co-transfected with the miR-384-5p mimic and the wild-type PGC1β 3′-UTR target sequence expression plasmid, the expression of the reporter gene was dramatically reduced. In contrast, the mutated version of the PGC1β 3′-UTR plasmid failed to respond to the miR-384-5p mimic. The findings indicated that miR-384-5p interacted with the PGC1β 3′-UTR and subsequently inhibited the post-transcriptional processing of PGC1β, such as stability of mRNA and translation. PGC1β is known to exert an anti-inflammatory effect in gout, downregulate macrophage inflammatory responses, and polarize macrophages to a less inflammatory state.23,25 We then sought to investigate the changes in the levels of PGC1β expressed in C. albicans-infected macrophages, macrophages transfected with the miR-384-5p mimic, or co-treated with C. albicans and miR-384-5p inhibitor. The expression of PGC1β was suppressed by C. albicans infection and miR-384-5p mimic solely, and upregulated by miR-384-5p inhibitor in C. albicans-infected macrophages. Macrophages with forced overexpression of PGC1β produced fewer pro-inflammatory cytokines than the controls. These results suggest that the positive effect of miR-384-5p on C. albicans-induced ALI is at least partially mediated by suppressing PGC1β.
Activation of inflammatory signaling pathways is one of the mechanisms by which C. albicans induces ALI. 30 C. albicans can activate the NF-κB, MAPK, Akt, and STAT3 pathways. Manno-proteins derived from C. albicans are ligands for toll-like receptor 4 (TLR4) in C. albicans infection.30,42,43 NF-κB is a downstream signaling pathway of the TLR-4 pathway. C. albicans induced inflammatory responses partially through the NF-κB signaling pathway. MAPK and Akt are two other signaling pathways involved in C. albicans-induced ALI, which modulate TLR-4-mediated inflammation in ALI. 30 The STAT3 signaling pathway is important for the differentiation and maturation of Th17 cells and the IL-17 secretion, which is particularly implicated in the host defense against C. albicans infection.44–46 In our study, it was found that targeting C. albicans-upregulated miR-384-5p significantly inhibited the activation of the NF-κB, MAPK, and Akt signaling pathways triggered by C. albicans but not the STAT3 pathway. These results suggest that miR-384-5p might be involved in TLR4-mediated signaling activated by C. albicans but unlikely to participate in regulating the function of Th17 cells and the secretion of IL-17. Further studies are required to explore the mechanisms underlying the modulation of TLR-4-mediated signaling by miR-384-5p in C. albicans infection.
Human mature miR-384 sequence is highly identical to that of mouse mature miR-384-3p, as shown in miRDB database. Given that miR-384-3p and miR-384-5p derive from the same premature form of mouse miR-384, we speculate that miR-384-5p is likely to be expressed in human tissues under certain conditions but not detectable under normal condition. Moreover, miR-384-3p might function similarly in human tissue under pathological conditions as miR-384-5p in the mouse model. Further studies are needed to address these questions and investigate the association between the function of miR-384-5p and its clinical significance.
In conclusion, the present study demonstrates a positive role of miR-384-5p in C. albicans-induced ALI. Our findings revealed that miR-384-5p is involved in inflammation caused by C. albicans infection in ALI by suppressing PGC1β and participating in the activation of the NF-κB, MAPK, and Akt signaling pathways induced by C. albicans.
Footnotes
Author contributions
Study design: DY and CX; Research performance: DY, CX, HT and AY; Data collection, analysis and interpretation: DY, CX and LW; Manuscript preparation: DY, CX and LW.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the Department of Clinical Laboratory Sciences and the Department of Dermatology at the First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China.