
Abstract
Sickle cell disease (SCD) is an inherited blood disorder most common among African American and Hispanic American persons. The disease can cause substantial, long-term, and costly health problems, including infections, stroke, and kidney failure, many of which can reduce life expectancy. Disparities in receiving health care among African Americans and other racial/ethnic minority groups in the United States are well known and directly related to poor outcomes associated with SCD. As an orphan disease—one that affects <200 000 persons nationwide—SCD does not receive the research funding and pharmaceutical investment directed to other orphan diseases. For example, cystic fibrosis affects fewer than half the number of persons but receives 3.5 times the funding from the National Institutes of Health and 440 times the funding from national foundations. In this review, we discuss the health inequities affecting persons with SCD, describe programs intended to improve their care, and identify actions that could be taken to further reduce these inequities, improve care, control treatment costs, and ease the burden of disease.
Sickle cell disease (SCD) affects about 100 000 persons in the United States (Figure 1A). 1 The disease causes red blood cells to lose their normal disc shape and become sickle shaped and rigid. These sickle-shaped cells adhere to vascular walls, impede blood flow and oxygenation, and cause episodes of intense pain and other complications that affect multiple organ systems. Persons living with SCD often have chronic kidney disease, stroke, and liver and lung complications. 2 The life span of those with the most severe form of SCD is on average 30 years shorter than for the general US population (Figure 2). 3 -6 Although approximately 90% of persons with a diagnosis of SCD in the United States are African American, the disease also occurs among Hispanic American persons and persons of Mediterranean, Middle Eastern, and Indian descent. 2,7
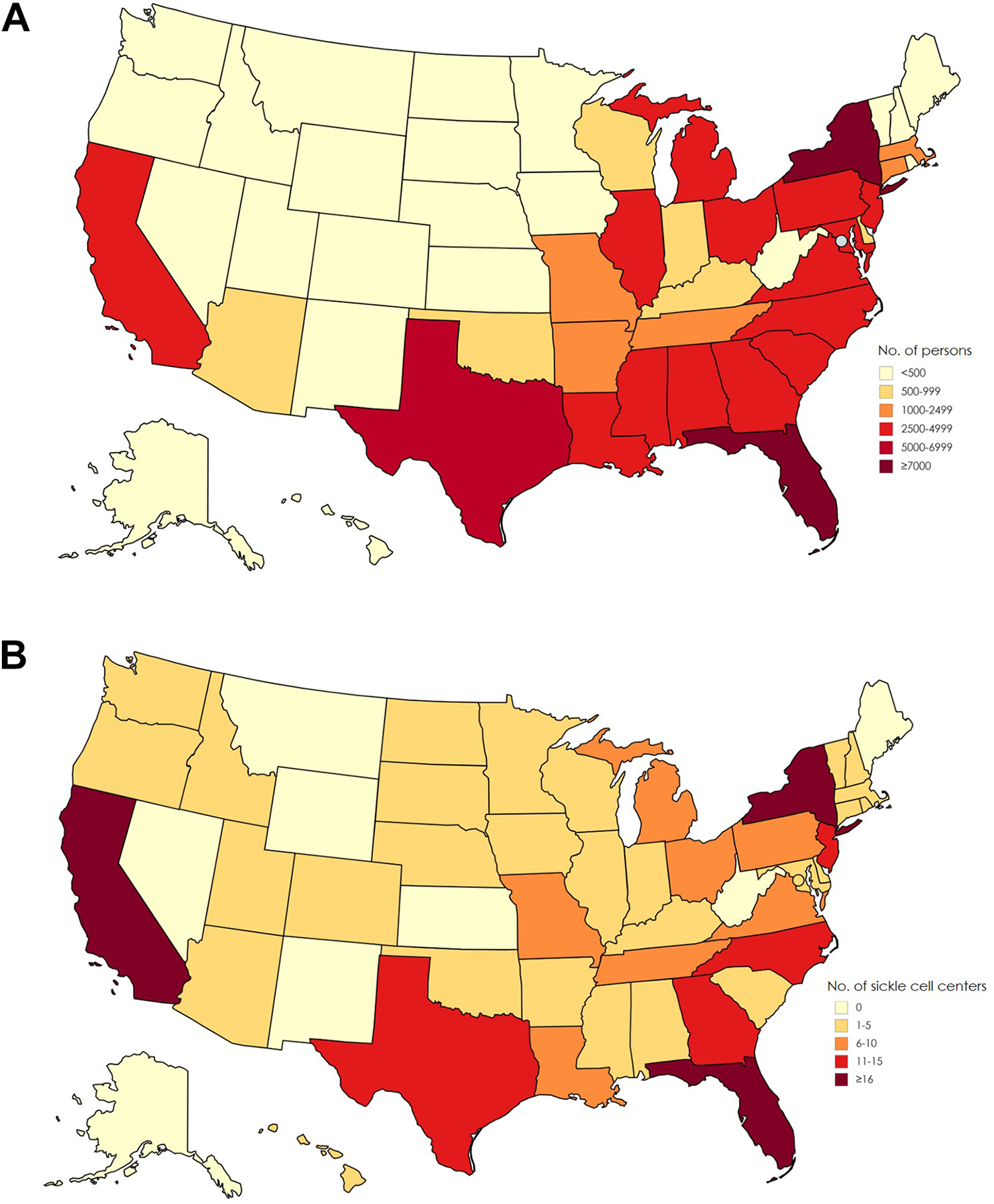
(A) Estimated number of persons with sickle cell disease, based on state-specific African American and Hispanic birth-cohort disease prevalence and 2008 US Census population, corrected for early mortality. Map previously published by Hassell 1 and reprinted with permission from the American College of Preventive Medicine. (B) Number of sickle cell disease treatment centers per state, 2017. Data source: Centers for Disease Control and Prevention (CDC). 17 Used with permission from CDC.
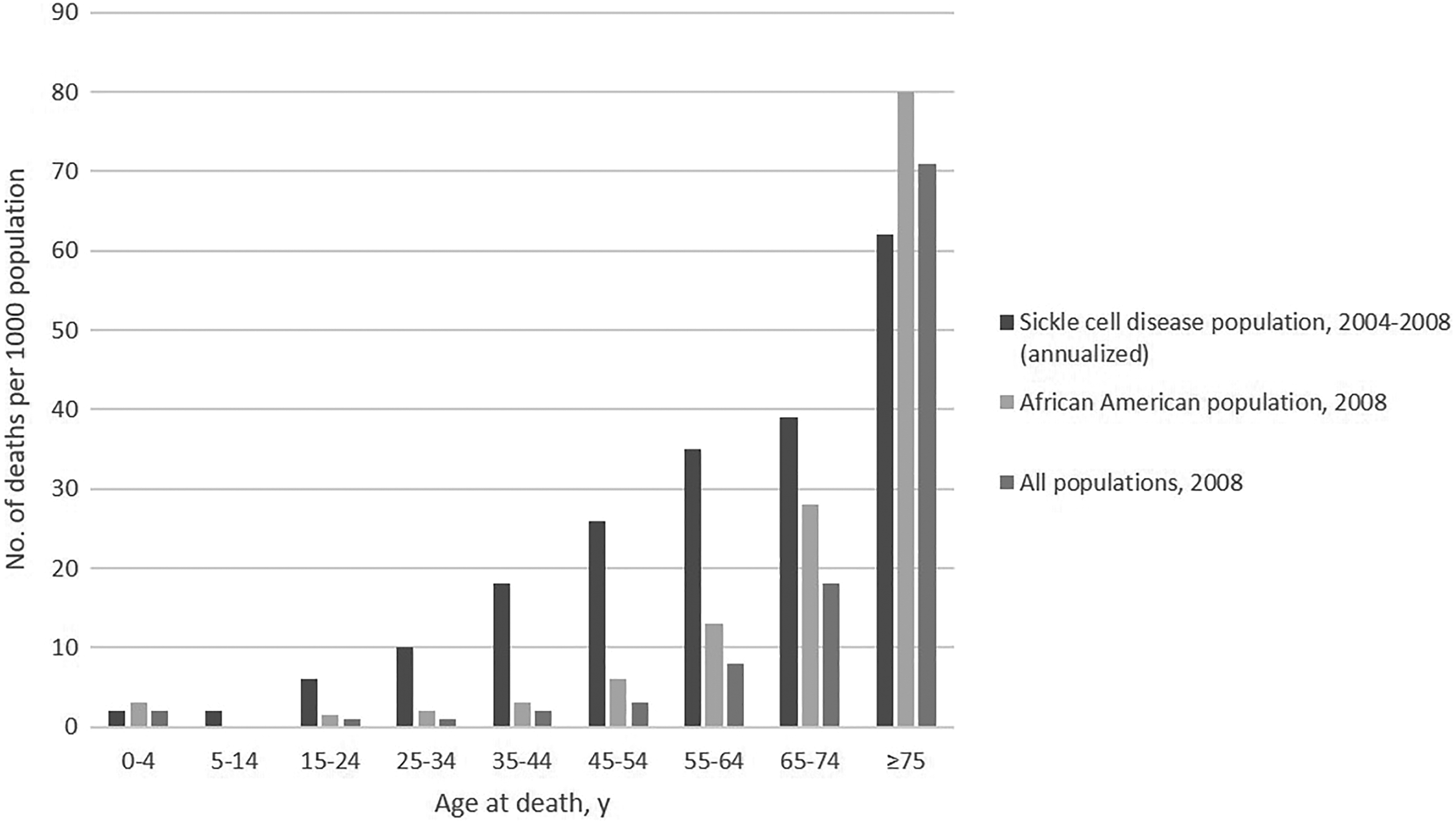
All-cause mortality rates for sickle cell disease identified through population-based surveillance (2004-2008), the African American population (2008), and all populations (2008), in California and Georgia. Source: Paulukonis et al. 6
As an orphan disease—one that affects <200 000 persons nationwide—SCD does not receive the research funding and pharmaceutical investment given to diseases that have a higher public profile or that have captured the attention of the medical research and funding communities. 8 As such, persons living with SCD usually have less access to comprehensive care and more difficulty in obtaining medication for pain relief than do persons with other chronic diseases. Most persons living with SCD are publicly insured, and Medicaid recipients with the disease have substantial barriers to receiving necessary services. Despite the low prevalence of SCD, the cost associated with managing the disease is high, and the disease has long-term complications. Health care providers with expertise in the treatment of SCD are in short supply, and disease-modifying treatments are limited. Here, we document disparities in health care access and quality of care for persons living with SCD and identify actions for reducing these disparities.
Disparities in Access to and Quality of Care for Persons Living With SCD
Lack of Access to Health Care and Specialty Care
A major challenge for persons with SCD is poor access to appropriate health care. 9 Although hemophilia and cystic fibrosis each affect fewer than half the number of persons in the United States than does SCD, 10,11 persons with these diseases benefit from more than 130 comprehensive treatment centers nationwide. These centers have multidisciplinary teams dedicated to improving health outcomes, providing quality care, and reducing costs and disparities in access to care for these patients. 12,13 In contrast, specialized SCD health care providers with comprehensive expertise are scarce, particularly in low-income and rural communities with limited resources (Figure 1B). 14 -17 Furthermore, in nonspecialized hospitals with few SCD patient populations, poor outcomes were reported, likely because of a lack of hematologists with SCD expertise. 18 A survey of 30 pediatric SCD centers found that only 18 centers routinely transitioned their patients to a hematologist specializing in adults with SCD upon aging out of the pediatric care system. 19
Poor Disease Outcomes
Disparities in life span exist between persons with SCD and the general US population. The median age at death for persons with SCD is 45, 25-30 years younger than that of the general US population. 3,20,21 Survival rates for children aged <18 with SCD have improved during the past several decades; a 2010 study found that 94% of persons with SCD survive to adulthood, an improvement from a 2004 estimated survival rate of 86% for children with SCD. 20 However, mortality rates were higher among young adults aged 20-24 from 1999 to 2009 than they were during the previous 2 decades. 3 The 2009 mortality rate for persons with SCD was 0.6 per 100 000 population among adolescents aged 15-19 but 1.4 per 100 000 population among young adults aged 20-24. Thus, mortality rates for young adults with SCD increased 2.3-fold as they transitioned from pediatric care to adult care. 3,22 This increase could partially be driven by limited access to specialized care in the adult setting, but more quality improvement efforts should examine how to address this disparity.
Rates of preventive care are also lower among patients with SCD than among patients with other chronic diseases. Newborn screening for SCD, which was implemented nationwide in 2006, decreased mortality among newborns and increased access to necessary specialized care. However, a survey of 39 state screening programs found that 12 programs did not follow patients long term after SCD diagnosis. 23 A comparison of counseling and other preventive services for adolescents at cystic fibrosis and SCD treatment centers found that 86% of adolescents with cystic fibrosis had received at least 1 immunization in the previous 2 years, whereas only 51% of adolescents with SCD had done so. 24 Most children with SCD are not monitored for stroke risk with transcranial Doppler ultrasound. 25 In a 2015 study of 4887 children aged 2-16 years with SCD, the annual rate of transcranial screening varied considerably among states, from 7% in Texas to 54% in South Carolina. 26 Finally, among 2821 children aged 3 months to 5 years with SCD, only 18% (range, 6%-27%) received a recommended 300 or more days of antibiotic prophylaxis, 27 and more than 25% had no documentation of having received recommended pneumococcal vaccinations. 25 Without these services, children with SCD are at greater risk of death and cognitive defects from infection and stroke than children with SCD receiving routine screening and preventive treatment. 28
Inadequate Access to Specialized Care
A 2010 analysis of 21 112 persons with SCD found that 60% used Medicare or Medicaid as their primary payer. 29 In contrast, from 2005 to 2009, only 32% to 38% of 13 100 persons with hemophilia and 22% to 32% of 4442 persons with von Willebrand disease expected to use Medicare or Medicaid as their primary payer. 30 These differences are important because Medicaid limits access to specialized health care. A retrospective claims analysis found that significantly fewer persons with Medicaid than with commercial insurance had visited a hematologist during the past year (Medicaid, 7% of 5612 vs commercial insurance, 43% of 2884; P < .001). 31 These disparities likely contribute to the difference in outcomes between persons with SCD and persons with other chronic conditions. 11,30 The proportion of uninsured adults with SCD is higher than that of children, which further compounds issues related to health care access. 32
Lack of Primary Care Providers
Persons with SCD, particularly young adults, often visit the emergency department (ED) or hospital for SCD-related concerns, possibly because of a lack of primary care providers specializing in SCD. 33 In 2014, approximately 250 000 ED visits and 90 000 hospitalizations in the United States were for SCD. In contrast, that same year, approximately 3500 ED visits and 2100 hospitalizations were for cystic fibrosis. 34 Furthermore, unaddressed symptoms of SCD and not having a primary care provider are associated with high rates of readmission. 35 The 30-day and 14-day readmission rates for patients with SCD were reported to be 33% and 22%, respectively, with approximately 80% of all readmissions to the same hospital. 29 SCD was reported as the fifth most common discharge diagnosis for Medicaid “superusers” (those with ≥4 hospital visits per year) aged <65. 36,37
High rates of hospitalization, ED visits, and hospital readmissions contribute to high treatment costs for SCD. In the United States, the annual cost to treat persons with SCD was more than $1.1 billion in a 2009 analysis of Medicaid data. 38 The total lifetime health care cost for a person with SCD is approximately $9 million, with estimated annual costs ranging from $35 500 to $112 000 for children aged <18 and $231 000 for adults. 39 Annual per-person costs for hemophilia ranged from $17 800 to $35 000 for persons using Medicaid and from $12 500 to $17 000 for persons using commercial insurance in 2016. 40
Barriers to Obtaining Pain Treatment
Persons with SCD have painful vaso-occlusive crises, diffuse inflammation, chronic organ damage, and chronic pain. 41 The 2014 National Heart, Lung, and Blood Institute expert panel report recommended opioid analgesia for severe SCD pain. 28 However, persons with SCD are often stigmatized when seeking pain relief. A 2005 survey of 109 physicians treating patients with SCD revealed that many physicians believe that attitudes toward opioid addiction may result in undertreatment of pain and “a disbelief in patient’s report of pain severity.” 42 Furthermore, a 2014 study found that nurses’ beliefs about the extent of opioid abuse and addiction among patients with SCD were substantially more negative than those of physicians. 43 Measures to manage the opioid epidemic also limit access to necessary pain relief for these patients. In 2017, the US Drug Enforcement Administration and CVS Health issued policies to reduce opioid production and limit prescriptions, with exemptions for patients with cancer or in palliative or end-of-life care but no accommodations for patients with SCD. 44 -47 However, in 2019, the Centers for Medicare & Medicaid Services released a final policy recommending that Medicare beneficiaries with SCD be exempt from these opioid safety restrictions. 48 Nevertheless, because of the growing opioid epidemic, access issues remain, and state Medicaid offices should provide similar exemptions.
The ED presents an additional barrier for patients with SCD, who frequently have long wait times when seeking treatment for pain. A multicenter retrospective study of EDs found that patients with SCD experiencing episodes of acute pain waited 70-75 minutes longer on average than guideline recommendations to receive analgesics. 49 Another national survey reported that patients with SCD waited 50% longer to see an ED physician than did patients with long-bone fractures, despite adjusting for race and triage priority. 50,51 Delays in treating persons with acute SCD pain have consequences: tissue ischemia and inflammation occur during the early prodromal phase, and aggressive pain treatment during this phase may reduce the duration of pain, prevent complications, and avert irreversible tissue damage. 51,52
Insufficient Disease-Modifying Treatments
Treatment options for persons with SCD are limited despite continued improvement in the understanding of SCD pathophysiology. Hydroxyurea and Endari (
SCD research is also underfunded. A 2013 analysis comparing funding for SCD with funding for cystic fibrosis (the latter of which affects one-third as many persons as SCD) found that research funding from the National Institutes of Health was 3.5-fold higher and funding from national foundations was 440-fold higher for cystic fibrosis than for SCD. 58 Higher funding for cystic fibrosis has likely contributed to numerous therapies receiving FDA approval during the past decade, whereas only 1 drug was approved for SCD during the same period. 58,59
Successful Strategies for Treating Orphan Diseases
Several strategies for treating other orphan diseases could reduce the health care disparities associated with SCD; in particular, a national surveillance system, enhanced models of access to care, and an increase in the number of trained providers.
A National Surveillance System
National surveillance systems can help determine where to allocate health system resources and what gaps exist in clinical research. The ability to accurately determine the benefits of coordinated, comprehensive care and to assess ongoing needs depends on the availability of surveillance data that can be used by researchers and policy makers to identify the population affected by SCD and to measure health outcomes, cost-effectiveness, and quality of life. 60
National surveillance programs for other chronic diseases, such as amyotrophic lateral sclerosis or hemophilia, can provide effective models or benchmarks for SCD. 61,62 The Centers for Disease Control and Prevention (CDC) supports the Sickle Cell Data Collection program in 2 states (California and Georgia) to collect, synthesize, and disseminate multisource, population-based, longitudinal data on SCD. 63 The American Society of Hematology and the Sickle Cell Disease Association of America support increasing funding for national SCD surveillance and expanding CDC’s population-based surveillance work to other states. 64 As a result of these advocacy efforts, in 2019 CDC received a 1-year funding increase of $1 million for SCD surveillance. 61,62 However, for a surveillance program to be truly effective, sustained funding is required for the existing system, and additional congressional funding is needed to include more states and ensure broader participation. 63,65
Improving Access-to-Care Models
A substantial proportion of persons with SCD receive services through Medicaid; thus, the Centers for Medicare & Medicaid Services is important in improving access to health care. Better incentives to provide coordinated health care services for Medicaid recipients are needed. 9 To encourage funding increases, payers need to be educated on the cost-effectiveness of coordinated, comprehensive care delivered through integrated treatment centers. Coordinated care models would likely reduce the high rate of ED visits among this population. Models such as a day clinic for SCD care have already substantially reduced both the number of ED visits and hospitalizations by persons living with SCD. 18,66
Hemophilia treatment centers are a disease-specific network of institutions recognized for providing comprehensive patient care. In the United States, more than 130 centers serve 20 000 persons with hemophilia. 61,67 This network is financed by several government agencies, including the US Public Health Service’s outpatient program, which generated revenue for maintaining and expanding services; the Health Resources and Services Administration, which funds $4.9 million to provide comprehensive services not covered by insurance; and CDC, which funds $5 million for research, surveillance, and prevention activities. 68,69 Multiple government funding mechanisms should be explored to expand the number of treatment centers for SCD. The Sickle Cell Treatment Act of 2003 (S. 874 and H.R. 1736) expanded funding for best-practice education and specialized treatment programs in the 40 SCD treatment centers nationwide. 70 However, with at least 100 000 persons with SCD living in the United States, the number of such centers would need to be increased to achieve the same level of resources and coordinated care as that available for hemophilia. 71
The Health Resources and Services Administration is important for improving treatment practices for SCD through the Sickle Cell Disease Treatment Demonstration Program, 72 a federally mandated and funded program that aims to establish models for SCD prevention and treatment by coordinating service delivery, genetic counseling and testing, bundling of technical services, and provider training. Currently, this program does not cover the southern US states, where most patients with SCD reside. 73,74 To meet these congressionally prescribed goals, the program needs increased federal funding.
Community health centers are central to public health and provide primary care, prenatal care, dental services, and health education to underserved and uninsured populations. For example, the Ryan White Comprehensive AIDS Resources Emergency Act of 1990 substantially improved outcomes for low-income and underserved persons with AIDS. 75 However, community health services require additional funding to address the complex challenges associated with SCD to improve patient outcomes, similar to the center-based initiatives that were implemented for AIDS. Community health centers risk losing 70% of federal funding should Congress not reauthorize financial support by September 2019, which would reduce services to uninsured and low-income persons with SCD.
Community health workers who specialize in treating persons with SCD provide social support, offer informal counseling, and aid in navigating health systems. 25 The Sickle Cell Disease Association of America offers a certification program to expand the number of health workers qualified to support patients with SCD and has trained more than 300 health care workers (S. Fitzgerald, Community Engagement Manager at the Sickle Cell Disease Association of America, oral communication, July 2019). 75 Additional stable funding will be necessary for the program to be robust and sustainable to meet the needs of persons with SCD nationally. 76
The Health Resources and Services Administration also improves support for community-based organizations serving persons with SCD by funding the Sickle Cell Disease Newborn Screening Follow-up Program. This program aims to eliminate barriers to care by ensuring that persons with SCD diagnosed through the screening program receive follow-up counseling, education, help with access to a medical home, and other support services provided by community-based organizations. However, to identify, educate, and connect more persons to care services, additional funding is required to expand this program beyond the 15 states currently funded. 76
Addressing the Shortage of Providers
Most clinical fellowship programs for hematology are solely focused on hematologic malignancies, and only 2 programs allow for specialization solely within benign hematology. To address the limited number of providers with expertise in treating persons with SCD, the American Society of Hematology (hereinafter, the Society) has implemented several initiatives, including adding SCD modules to the benign hematology curriculum and sponsoring the Sickle Cell Disease Centers Workshop on adults with SCD. 16,77,78
Primary care providers are a key target of the Society’s activities, given the number of patients they treat. 79 The Society also undertook a multifaceted initiative to address the burden of SCD, including developing new clinical practice guidelines, planned for publication in 2019, and increasing access to care (including expanding the surveillance program).
Project ECHO (Extension for Community Healthcare Outcomes) connects clinicians with expertise in complex disorders, such as SCD, with local clinicians through telementoring. 80 The Health Resources and Services Administration is using Project ECHO in the Northeast, Southeast, Midwest, Heartland, and Southwest to allow clinicians to connect with experts to increase their knowledge about managing patients with SCD. 81 Thus, Project ECHO could address the shortage of SCD providers by expanding the reach of specialists.
Public Health Implications
Morbidity among persons with SCD is high; having the disease results in decreased quality of life and premature death. 2 In addition, persons with the disease may be stigmatized by institutions, health care providers, and the general public because of opioid use and sociodemographic characteristics. 82 The lack of a national surveillance program prevents extensive and accurate epidemiologic research, which likely confounds accurate estimates of treatment outcomes and health care use. 1 Access to qualified health care is limited by a lack of providers with expertise in SCD, insufficient numbers of (and funding for) integrated comprehensive care programs for adults, and the stigmatization of pain relief for SCD. These factors result in a costly dependence on acute care from hospitals and EDs, illustrating the need to improve community care with better funding of community health centers, community health workers, and care coordination services, such as those offered by community-based organizations. The reliance of patients with SCD on EDs disrupts continuity of care and reflects a need to shift disease management processes from an acute care model to a chronic care model, with an increased emphasis on preventing pain episodes and chronic complications. Furthermore, treatment options are limited.
Improving national surveillance, access to care, and targeted education of providers in areas of high SCD prevalence can be addressed in the near term. Increasing the pool of providers with expertise in SCD and finding new disease-modifying therapies are long-term goals requiring continued investment. Increasing the number of comprehensive care centers and regionalization of care will likely improve outcomes. 18
Greater disease-specific focus and funding is needed for research, surveillance, education, and care delivery to improve the health outcomes and well-being of this underserved population.
Footnotes
Acknowledgments
The National Minority Quality Forum is a nonprofit, nonpartisan, Washington, DC–based health care research and education organization founded in 1998. This review is based on issues raised at the 2017 Forum’s Summit on Sickle Cell Disease. The authors acknowledge Mary Hulihan at the Centers for Disease Control and Prevention for her guidance in preparing this article. Medical writing and editorial support were provided by Arjun Menon, PhD, Healthcare Consultancy Group.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Medical writing and editorial support were provided by Arjun Menon, PhD, Healthcare Consultancy Group, which was funded by Global Blood Therapeutics, Inc, South San Francisco, California.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.