
Abstract
CRISPR-mediated genome editing has undoubtedly revolutionized genetic engineering of animals. With the ability for virtually unlimited modification of almost any genome it is easy to forget which amazing discoveries paved the way for this ground-breaking technology. Here, we summarize the history of genome editing platforms, starting from enhanced integration of foreign DNA by meganuclease-mediated double-strand breaks to CRISPR/Cas9, the leading technology to date, and its re-engineered variants.
Introduction
Genetic engineering of organisms is the key to biomedical and biotechnological research. The ultimate tool would allow precise and unlimited modification of any given nucleic acid sequence in a simple fashion. With the rise of genome editing, we have never been closer to accomplishing this goal. This review will summarize the exciting key developments leading to the breakthrough of genome editing with a particular focus on CRISPR-based tools as the leading technology to date.
Genetic engineering started with transgenesis, the deliberate transfer of genetic information from one organism to another. In 1974, Rudolf Jaenisch demonstrated that viral DNA injected into blastocysts led to stable integration in the genome of mice. 1 This is considered to be the first transgenic animal generated. The term ‘transgenic’ though was introduced only in 1981 by Gordon and Ruddle when they and others showed that injection of DNA into pronuclei of murine zygotes conferred stable germline transmission of the genetic modification to the next generation.2–5 Although integration occurred randomly and without copy number control, researchers were able to establish a stable genetically engineered mouse line for the first time. At the same time, two groups independently isolated pluripotent mouse embryonic stem cells (ESC)6,7 which were able to colonize the entire embryo, including the germ line, upon injection into host blastocysts. 8 In these cells, Oliver Smithies and Mario Capecchi employed the process of homologous recombination to inactivate a specific gene in the mouse.9,10 This ground-breaking discovery, termed gene targeting, allowed site-directed modification of the genome for the first time and earned Mario Capecchi, Martin Evans and Oliver Smithies the 2007 Nobel Prize in Physiology or Medicine. By the additional coupling with a site-specific recombination system (i.e. Cre-lox), the group of Klaus Rajewsky expanded the scope of gene targeting in a time and cell type specific fashion and this approach soon became the gold standard for genome engineering in mice for the next decades.11,12 Despite its undeniable power, the homologous recombination approach exhibited several limitations. Firstly, the poor integration frequency at a specific site in zygotes required the time-consuming detour though ESC targeting and subsequent injection into blastocyst for the generation of genetically modified (GM) mice. 13 Secondly, due to the low integration frequency, a selectable marker (often an antibiotic resistance gene) was required to determine which ESCs integrated the DNA. The use of this marker would leave undesired genetic material that required subsequent removal to avoid altering expression of the gene.9,10 Finally, the approach was largely limited to mice due to the lack of protocols for ESC isolation or the failure to isolate such cells in most other species. 14
The rise of genome editing (1985–2013)
How could these limitations be overcome? One important step was the discovery that double-strand breaks (DSB) dramatically enhance the integration of foreign DNA in mammalian cells.15–17 These studies employed the meganuclease I-SceI to induce DSB in the genome of mouse cells. DSBs subsequently recruit the endogenous repair machinery and are either repaired by end-joining pathways, which can result in random insertions and deletions (INDELs) or, if a DNA repair template is provided, by homology-directed repair (HDR) pathways (Figure 1). Meganucleases are low frequency cutters with a recognition sequence of typically 20-30 bp discovered in yeast in 1985 (Figure 2). 18 They are involved in gene conversion processes like duplication and have now been identified in many microbial genomes. 19 Although hundreds of naturally occurring meganucleases exist, their fixed recognition sequence drastically limited the choice of targetable loci. Subsequent re-engineering of the DNA recognition site led to more flexibility and enabled site-directed genome modification in mammalian cells, mice and rats.20–22 Yet, reconfiguring the structure of an entire protein for every single target site was tedious.
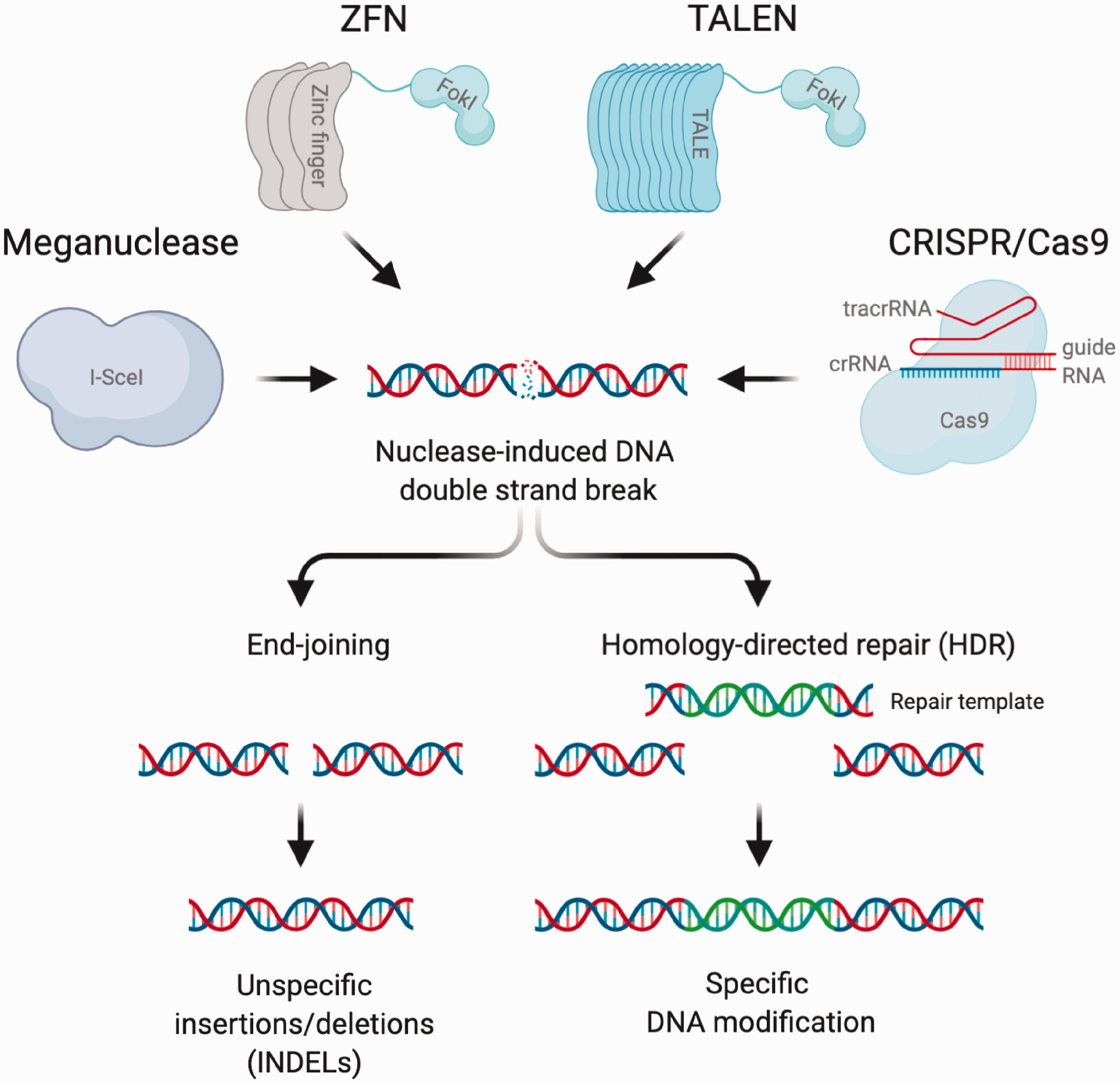
Mechanism of major genome editing platforms. All four major genome editing systems lead to double-strand breaks (DSBs) at the desired locus. These DSBs breaks are either repaired by error-prone end-joining pathways resulting in random insertions and deletions (INDELs) or, if a DNA repair template is provided, by homology-directed repair (HDR). The latter mechanism can be exploited to introduce site-specific mutations into the locus of interest. While meganucleases, zinc finger nucleases (ZFN) and transcription activator-like effector nucleases (TALENs) rely on protein-DNA interaction for target recognition CRISPR/Cas9 utilizes a dual guide RNA composed of a generic tracrRNA and a specific crRNA.
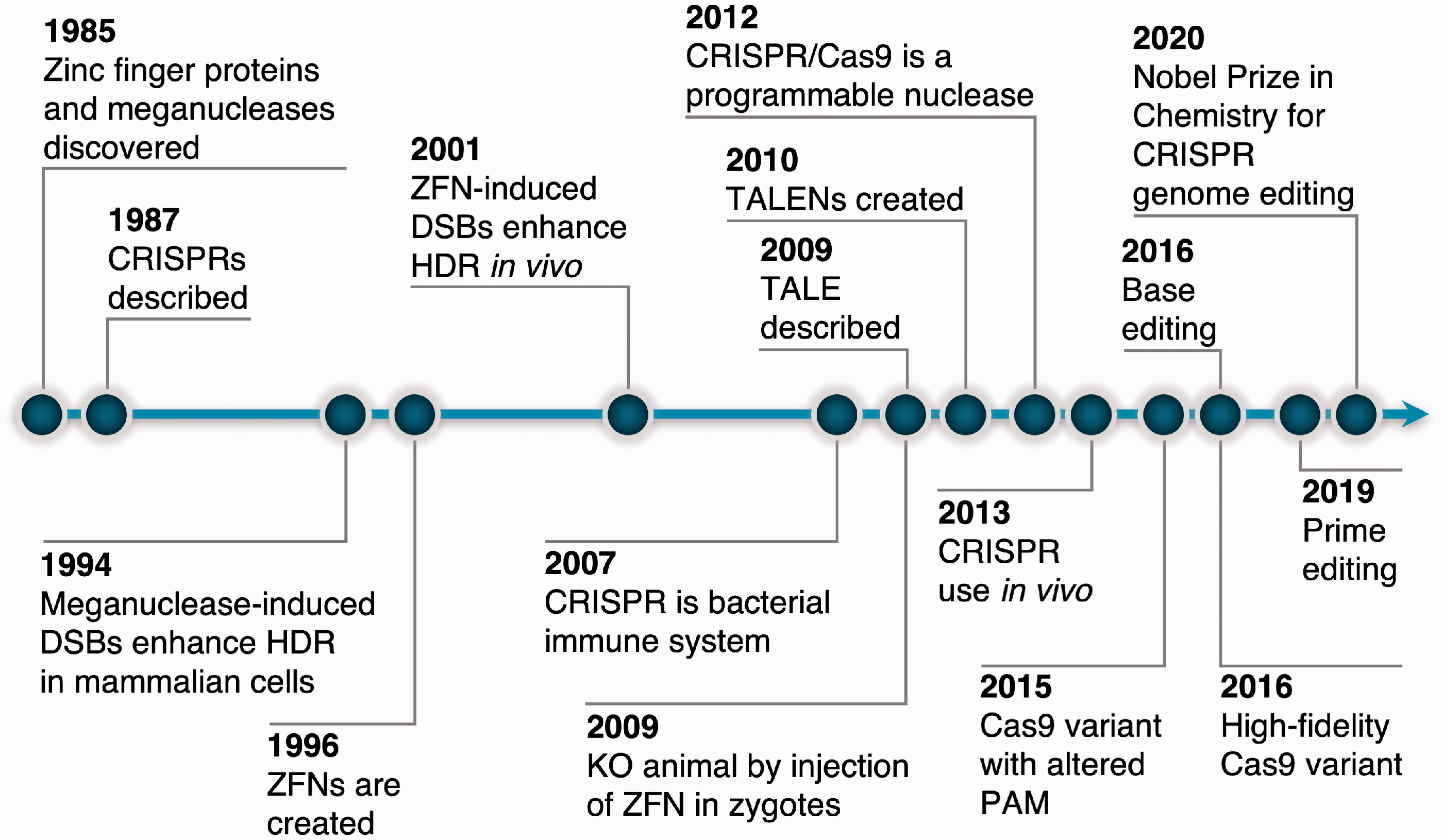
Key developments in the history of genome editing. DSBs, double-strand breaks; HDR, homology-directed repair; KO, knock-out; PAM, protospacer adjacent motif; TALENs, transcription activator-like effector nucleases; ZFN, zinc finger nucleases.
What if there was a protein motif which has already naturally evolved to bind various different nucleotide sequences? Zinc fingers are small protein motifs with sequence specific DNA binding capacity. They were first discovered in 1985 as part of a transcription factor in frog oocytes but are in fact present in many species, including humans.23,24 They were termed zinc fingers as they are stabilized by a zinc ion in order to bind their DNA target. In contrast to other DNA binding motifs, they utilize a cascade of modules in which a few key residues in the protein mediate recognition. Each module harbours a DNA binding motif specific to three consecutive base pairs of the target sequence. This modular structure makes them highly versatile in binding specific DNA sequences of various lengths. 24 In 1996, researchers fused these zinc finger modules to the DNA cleavage domain of the restriction enzyme FokI in order to generate a programmable nuclease, termed zinc finger nucleases (ZFN). 25 The ease of inducting DSB by ZFN subsequently led to a number of applications in biotechnology and biomedicine. Pilot experiments in 2001 in Xenopus oocytes demonstrated the feasibility of enhanced HDR by ZFN. 26 Shortly thereafter, the approach was optimized and applied to other organisms, like Drosophila and human cells, and also employed therapeutically to correct the human SCID mutation in vitro.27–29 The latter study introduced the term ‘genome editing’ in 2005 for the first time as an analogy to word processing.29,30
DSB, like those caused by ZFNs, can be repaired by HDR if a repair template is provided. That was how knock-out (KO) and knock-in (KI) mutations have been generated with genome editing. However, most DSB in vertebrate cells are repaired by error-prone end-joining pathways which frequently result in INDELs and not by HDR. Inspired by previous work in lower organisms such as flies, worms and plants, in 2008 three independent groups realized that one can utilize the thus far undesired INDEL mechanism to efficiently induce KOs in zebrafish and mammalian cells without the need of HDR with a repair template. This process was so efficient that no selection was needed.31–33 A break-through for the generation of GM mammals came in 2009 when it was shown that microinjection of DNA or mRNA encoding for specific ZFNs in rat zygotes produced the first KO rats. 34 This technology was swiftly adapted to generate GM model organisms like mice and rabbits but also livestock animals such as cattle and pigs (see Supplemental Table S1 for references).
Another major discovery in the field of designer nucleases arose with the construction of transcription activator-like effector nucleases (TALENs) in 2010.35,36 Researchers realized that the DNA binding domain of ZFN can be exchanged with more flexible and easier to generate DNA binding modules. These modules originated from TALE proteins discovered in plant pathogenic bacteria of the genus Xanthomonas. These bacteria utilize the sequence specific binding capacity of TALEs to manipulate the gene expression of the infected plant cell in their favour. Like zinc finger proteins TALEs possess a variable DNA binding domain. However, there is a simple code of recognition where one module in the protein corresponds to only one of the four nucleotides (A, T, C or G). Various combinations of these four modules can therefore target any DNA sequence. Furthermore, as only two amino acids are responsible for the nucleotide specificity, TALEs are easier to generate than are zinc finger proteins. 37 For genome editing, TALENs stem from the fusion of a sequence-tailored TALE DNA binding domain to the FokI endonuclease domain (analogous to that used for ZFN).35,36 As the design and the generation of TALENs required less effort and was more flexible, they immediately started to replace ZFN for genome editing. Soon after their first application in cell culture, TALENs were used to modify the genomes of a number of model organisms including worms, zebrafish, frogs, rats, mice and rabbits. But they were also employed in livestock such as pigs, goats, sheep and cattle as well as non-human primates (see Supplemental Table S1 for references).
The history of CRISPR genome editing (1987–2013)
What if there was a protein which has programmable DNA binding capabilities like TALE and zinc finger proteins but does not need time-consuming construction of a new protein domain? Ideally, this protein would already even exist as a nuclease in nature. Unknowingly, such a system had already been discovered, one we now know as the CRISPR/Cas system in bacteria.
In the late 1980s, repeats were observed in the genome of Escherichia coli which, unlike typical tandem repeats, were separated by non-repeating spacer sequences. 38 The nature of these repeats remained elusive for more than a decade until improved sequencing technologies enabled the decoding of many other genomes. In 2000, Mojica and co-workers realized that these repeats are present in many other bacteria and virtually all archaea, which pointed to an important role for these elements. 39 In addition, these repeats were shown to be associated with conserved genes subsequently called CRISPR-associated or Cas genes, which are in fact naturally occurring endonucleases. This was also the first use of the term CRISPR for clustered regularly interspaced short palindromic repeats. 40 In 2005, three research groups independently demonstrated that the spacer sequences are identical to genomes from phages and other foreign genetic elements.41–43 In 2007, the nature of CRISPR was finally proven, when Horvath and co-workers demonstrated that infected bacteria incorporate spacers derived from phage which in turn directs the Cas protein to the genome of the invaders where it precisely cuts the phage DNA. The CRISPR/Cas system therefore provides a defence mechanism against infections, similar to the adaptive immune response known from higher organisms such as mammals. 44 Soon after it became clear that so-called CRISPR RNA (crRNA) is transcribed from the spacer to guide the Cas protein to its target 45 and that the target is DNA. 46 A short sequence motif adjacent to the crRNA targeted sequence on the target DNA, termed the protospacer adjacent motif (PAM), was not only shown to be critical for cleavage but also responsible for self vs. non-self discrimination of the CRISPR system.47–50 In addition, it was shown that in a certain CRISPR system only a single Cas protein, that is Cas9, confers DNA cleavage49,51 and trans-activating crRNAs (tracrRNAs), which play a role in the maturation of crRNA, are critical for its activity. 52 Undoubtedly, the most important experiments were conducted independently by the group of Emmanuelle Charpentier in collaboration with Jennifer Doudna and almost in parallel by the group of Virginijus Siksnys. They demonstrated that the CRISPR/Cas9 system can be reconstituted in vitro and programmed to target desired sequences in other species which proved its applicability as a programmable nuclease for genome editing.49,53 Charpentier and Doudna, in conjunction with Martin Jinek, also simplified the system when they fused the crRNA and tracrRNA into a single guide RNA (sgRNA) which retained full activity. 49 Immediately thereafter, ground-breaking studies from the Doudna group in parallel with the group of Feng Zhang and George Church adapted the CRISPR/Cas9 system for invivo genome editing in mammalian cells.54–56 Soon after CRISPR/Cas9 was used by the Jaenisch group to generate mice with multiple KO, conditional KO and reporter genes by zygote injection.57,58
Thus, instead of constant target specific re-engineering of protein modules as needed for ZFNs and TALENs, the Cas9 endonuclease requires only the simple construction of a short RNA molecule for genome editing at a given locus. In addition to its simple low-cost design this approach can also swiftly be multiplexed to target multiple genes at the same time. As this system was easy to adapt by any biomedical laboratory, CRISPR genome editing revolutionized the field and subsequently Emmanuelle Charpentier and Jennifer Doudna were awarded the 2020 Nobel Prize in Chemistry for its discovery. To date, this technology has been used to modify the genome of virtually all model organisms including fly, worm, zebrafish, frogs, rat, rabbit, pig, cattle, goat, sheep and non-human primates (see Supplemental Table S1 for references).
Natural and engineered CRISPR systems (2013–present day)
The Cas9 of Streptococcus pyogenes (SpCas9) used in the original publication has a simple PAM requirement (NGG) and CRISPR/Cas9 is still by far the most popular programmable Cas nuclease system for genome editing. Nevertheless, researchers have constantly searched for other Cas9 or Cas9-like proteins and developed a number of engineered versions of Cas9 with different properties, such as alternative PAM requirements, protein size, or substrate specificity to harness them for genome editing. 59
Invivo mutagenesis of specific organs in adult animals as well as gene therapy in humans is most often accomplished by means of viral vectors. The large protein size of SpCas9 (about 1400 amino acids), however, does not allow for delivery using the popular adeno-associated virus system. In search for smaller Cas9 variants, researchers discovered in 2015 that Staphylococcus aureus utilizes a much smaller Cas9 (about 1000 amino acids). This allowed genome editing in specific organs in mice by viral delivery of CRISPR/Cas9. 60 Many other Cas proteins have followed which differ in their protein size, PAM requirement, spacer length and editing specificity. With Cas12a, initially termed Cpf1, an entirely new class of CRISPR proteins was introduced in 2015. 61 This system broadened the applicability of genome editing to TT rich PAM targets as Cas12a uses the PAM sequence TTTV (V equals A, G or C). Only one year later, it was shown to work in vivo by the production of KO mice.62,63 Previously inaccessible loci could now be edited with this system. Whereas all known CRISPR systems until this point targeted DNA, the group of Feng Zhang discovered the CRISPR/Cas13 system, an RNA editing system, in 2017. 64 By targeting RNA, CRISPR/Cas13 can be used to modify the function of a gene by degradation of the mRNA allowing the reduction of a genes function by knock-down instead of a complete removal. Cas13 orthologs have recently also been shown to be effective in vivo in several animal embryos including zebrafish and mice. 65
Apart from harnessing natural Cas proteins, researchers have started to re-engineer the well-characterized Cas9 to develop new variants. These efforts pursued mainly two goals: broadening the range of targetable site by changing PAM specificities and enhancing the fidelity of Cas9. One disadvantage of the CRISPR system are off-target modifications which represent undesired mutations at loci that resemble the on-target sequence. It has been shown that SpCas9 can tolerate several mismatches depending on their distribution and position in the guide sequence. 66 Off-target mutations during genome editing in animals with a short generation cycle like mice is undesirable but can be partially compensated by backcrossing to remove the unwanted mutation. In contrast, off-targets in long-lived animals, including most livestock, and especially during therapeutic intervention in humans must be avoided. Therefore, researchers are still researching for ways to increase the specificity of Cas9. In a first attempt, the Cas9 protein has been used as a tandem, much like ZFNs and TALENs before. To this end one of the two nuclease domains of Cas9 was inactivated leading to a nickase which cuts only one strand of DNA. This approach proved to increase the specificity of the system in vitro and in vivo in mice. 67 The first high-fidelity protein variant of Cas9 was generated three years later in 2016 by Feng Zhang’s group. They used structure-guided engineering in order to change residues in the protein to prevent non-specific DNA binding. 68 Currently, many more variants have been created which can discriminate even single base pair mismatches. 69 However, none of these versions seem to retain the on-target activity of wild-type SpCas9. 70 Another limitation of the CRISPR system for genome editing is the requirement of a PAM sequence like NGG for SpCas9. This limits the targeting scope of Cas9 resulting in inaccessibility of certain loci. The first Cas9 with an altered and simplified PAM to overcome this limitation was introduced in 2015 by J. Keith Joung’s group. 71 This work, performed in zebrafish and human cells, demonstrated that a re-engineered SpCas9 efficiently recognized alternative PAM sequences like NGA. To date re-engineered Cas9 variants can recognize a variety of PAM sequences. Most recently, a variant with near-PAMless activity was constructed although this increased flexibility unfortunately still affects on-target activity. 72
Another risk of genome editing with nucleases like Cas9 are the undesired effects of DNA DSBs. Although these DSBs facilitate the introduction of foreign DNA they can also lead to undesired DNA modifications and even re-arrangements within the genome which must be avoided, especially in long-lived animals and precision human medicine. To circumvent DNA DSBs, the group of David Liu introduced base editing in 2016. 73 Here, a catalytically inactivated Cas9 is fused to a deaminase enzyme domain. This system allows single-base pair conversion of specific nucleotides, like C to T, without a DSB and without relying on HDR using donor templates. Base editing has now been used in many model organisms such as zebrafish, frogs, mice, rats and rabbit but also in livestock for example pigs, goat and sheep, and non-human primates (see Supplemental Table S1 for references).
Base editors can only convert certain nucleotides and cannot introduce deletions or insertions. To overcome these limitations another exciting CRISPR application was introduced in 2019. 74 Prime editing makes use of a Cas9 nickase fused to a reverse transcriptase. A dual functional prime editor gRNA (pegRNA) guides the Cas9 nickase to the desired locus. In addition, the pegRNA also harbours the template for the reverse transcriptase to copy the intended genetic change into the target DNA, starting from a short primer binding site complementary to the second strand of the target DNA. This system can install specific INDELs and all base-to-base conversions without relying on HDR and DNA DSB. Initially developed in vitro, this system has already proven to be applicable to model organisms such as mice. 75 Prime editing holds great promises for precise modification in animals and has clinical potential for human health as it shows exceptionally high specificity.
Apart from genome editing, the specific DNA binding capacity of catalytically inactive Cas9 has also led to many other applications in vitro and in vivo by fusion of different effectors. This has enabled site-specific applications such as gene regulation with transcriptional effectors, modification of the epigenome by epigenetic modifiers and live cell chromatin imaging by fluorescent proteins. 59 In addition, CRISPR-based technologies have led to numerous other exciting applications, including in agriculture and healthcare, which are beyond the scope of this review. 76
Conclusions and future directions
Gene editing has revolutionized biomedical research. Starting from the realization that meganuclease-mediated DSBs enhance integration of foreign DNA, many researchers have searched for an optimal tool to introduce these breaks in the DNA in a sequence specific fashion. Protein engineering led to the use of programmable ZFNs and, shortly thereafter, TALENs, which soon paved the way to simple and robust RNA-guided CRISPR genome editing. Due to its superior applicability, CRISPR has quickly become routine in many biomedical laboratories. Furthermore, the predominantly used and still most efficient SpCas9 system has been re-engineered for higher specificity and widened range of targetable loci but also applications beyond genome editing. Additionally, many other CRISPR systems have been introduced for genome editing and even other applications.
Due to the high variability of bacteria and archaea, many other CRISPR systems are likely to be discovered in the future. These new variants hold great potential to overcome the current constraints of genome editing. The limitation of PAM availability is a common issue. 72 The discovery of new Cas proteins or intentional re-engineering of known variants without any PAM requirement but retained efficiency would simplify genome editing tremendously. Unintended mutations due to off-target effects are another main obstacle of current CRISPR systems used for genome editing. 77 Substantial efforts have already led to Cas9 variants with enhanced specificity and acceptable trade-off on efficacy. A high-fidelity Cas protein, natural or re-engineered, with retained efficiency has yet to be developed. 70
All genome editing tools only direct the endogenous repair machinery to a specific locus rather than directly inducing the desired genetic alteration. While off-target mutations can mostly be predicted due to a number of available algorithms, unintended alterations at the on-target locus are still largely unpredictable. Besides small INDELs large and/or complex genetic alterations have been shown to occur including chromosomal translocations and megabase-scale deletion. 78 Even newer generation tools aiming for high precision like prime and base editors are not entirely predictable in their outcome. 77 A consensus on criteria for quality control of genome edited animals will be essential in order to ensure reliable and reproducible research.
Even if applied with rigid quality control to reduce the above-mentioned challenges, the number of animals needed per experiment will likely not be reduced compared to ES cell mutagenesis. Moreover, the ease of use and widespread availability of genome editing tools could even lead to an increase in genetically engineered models generated and accordingly animals used in research. However, the power of genome editing lies in its potential to introduce mutations directly on desired genetic strain backgrounds and complex existing animal models. This largely avoids extensive animal breeding programmes to obtain required allele configurations to an extent unachievable by classical genetic engineering. In addition, the ability for precise alteration of alleles will reflect human diseases more precisely than previously possible and in species most appropriate to the research objectives, ideally being less sentient than classical models. This will help to apply genome editing in line with the concept of the 3Rs. 79
Supplemental Material
sj-pdf-1-lan-10.1177_0023677221994613 - Supplemental material for History of genome editing: From meganucleases to CRISPR
Supplemental material, sj-pdf-1-lan-10.1177_0023677221994613 for History of genome editing: From meganucleases to CRISPR by Simon E Tröder and Branko Zevnik in Laboratory Animals
Footnotes
Acknowledgement
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.