
Abstract
Since it was introduced 20 years ago, tamoxifen-inducible genetic recombination in vivo has become a standard tool in many fields. This technique has great utility, allowing precise temporal and spatial gene recombination mediated by expression of a Cre recombinase-oestrogen receptor hormone binding domain fusion protein. It is frequently used in developmental biology, either for accurate spatio-temporal gene deletion or for lineage-labelling. Administration of high doses of tamoxifen can rapidly induce abortion in pregnant mice but this can be partially overcome by progesterone co-administration. However, administration of tamoxifen to pregnant mice early in pregnancy may have potentially lethal effects on the mother independently of abortion, and can also severely perturb embryonic development. Despite this, only a few published studies mention this fact in passing, and standard parameters for successful or unsuccessful use of tamoxifen in pregnant mice have not been reported. Therefore, in the interests of providing a framework for more humane animal research, we describe our experiences of tamoxifen administration during early gestation in mice. These observations should assist the design of future studies in accordance with the principles of the three Rs (Replacement, Reduction and Refinement of Animals in Research).
Six female mice of the Gt(ROSA)26Sortm1(Kcnj11*V59M)Fmas mouse strain
1
were mated with male C57BL6/J mice (Charles River Laboratories, Margate, UK). Five to seven days after plugging, the pregnant females were injected subcutaneously into the scruff on one occasion with a single dose of 10–200 mg/kg tamoxifen (Sigma-Aldrich, Gillingham, UK) dissolved in corn oil (Sigma-Aldrich, Gillingham, UK; see Table 1 for study design). Tamoxifen is metabolized in vivo to 4-hydroxy-tamoxifen, which acts as an antagonist of the oestrogen receptor. In research, it can also be used to conditionally activate the Cre-ERT2 fusion protein to cause conditional genomic recombination. In this study, the mice did not carry a Cre-ERT2 allele, so tamoxifen treatment would have no effect on the maternal genome. These mice were derived from a colony that had been backcrossed for more than 10 generations onto the C57BL/6J background. They were housed in a specific-pathogen-free facility free from the major rodent pathogens except Helicobacter hepaticus, with a 12:12 h light:dark cycle, at 19–23℃, 55% ± 10% humidity, in individually-ventilated cages (Tecniplast UK Ltd, Rushden, UK) containing Grade 4 Aspen Chip bedding (Datesand Ltd, Manchester, UK), cardboard tunnels and Sizzle Pet nesting material (LBS Biotechnology, Horley, UK), with free access to Teklad 2916 mouse chow (Envigo, Huntingdon, UK) and tap water. Bedding was changed fortnightly, and animals were assessed daily for welfare. Injections took place in the morning in a flow hood, and mice were immediately returned to their home cage after injection. Unexpectedly, all (6/6) mice developed severe intrauterine haemorrhage 7–9 days post-injection, and 3/6 had to be euthanized as a humane endpoint for ethical reasons 1–3 days prior to the planned embryo harvest date on day E15.5 (Figure 1 and Table 1). The three animals euthanized early had been dosed with 200 mg/kg on E5.5, 100 mg/kg on E7.5 and 10 mg/kg on E5.5, respectively. This effect was apparent at a range of doses commonly used in the literature to activate the tamoxifen-inducible Cre recombinase (10–200 mg/kg). We compared the effects of unmated mice of the same strain and genotype injected subcutaneously with a single dose of 200 mg/kg tamoxifen. These mice were controls from another study. All control mice (104/104) survived without any deleterious effects up to 14 weeks post injection (Figure 1). This was highly statistically significant (p < 0.0001, two-tailed Fisher’s exact test comparing 6/6 injected pregnant mice with 0/104 injected non-pregnant mice). There is some evidence suggesting that tamoxifen dosage by oral gavage may be less toxic to pregnant mice.
2
We therefore treated a further two pregnant mice with 100 mg/kg tamoxifen by oral gavage on E5.5 or E7.5. Both mice also suffered intrauterine haemorrhage 8–9 days after treatment, with one of these being found dead the day before the planned embryo harvest date of E15.5 (p = 0.0002, two-tailed Fisher’s exact test comparing 2/2 gavaged mice with 0/104 control injected mice). Therefore, we conclude that even tamoxifen administered via oral gavage might be indirectly toxic to pregnant mice if given early in pregnancy.
Tamoxifen-injected pregnant mice started showing signs of severe distress 5–7 days post tamoxifen injection and were euthanized as a humane end-point. By contrast, tamoxifen-injected non-pregnant females showed no effects for at least 100 days post-injection. Deleterious effects including intrauterine haemorrhage in the mother and morphological defects in the embryo were seen in the majority of mice, irrespective of tamoxifen dose. Morphological defects included: necrotic embryos, pericardial oedema (PCO), cranial neural tube defects (cNTD) such as exencephaly, craniofacial defects (CFD) including cleft palate, subcutaneous oedema (SCO) and ophthalmic defects (Oph) including microphthalmia or anophthalmia.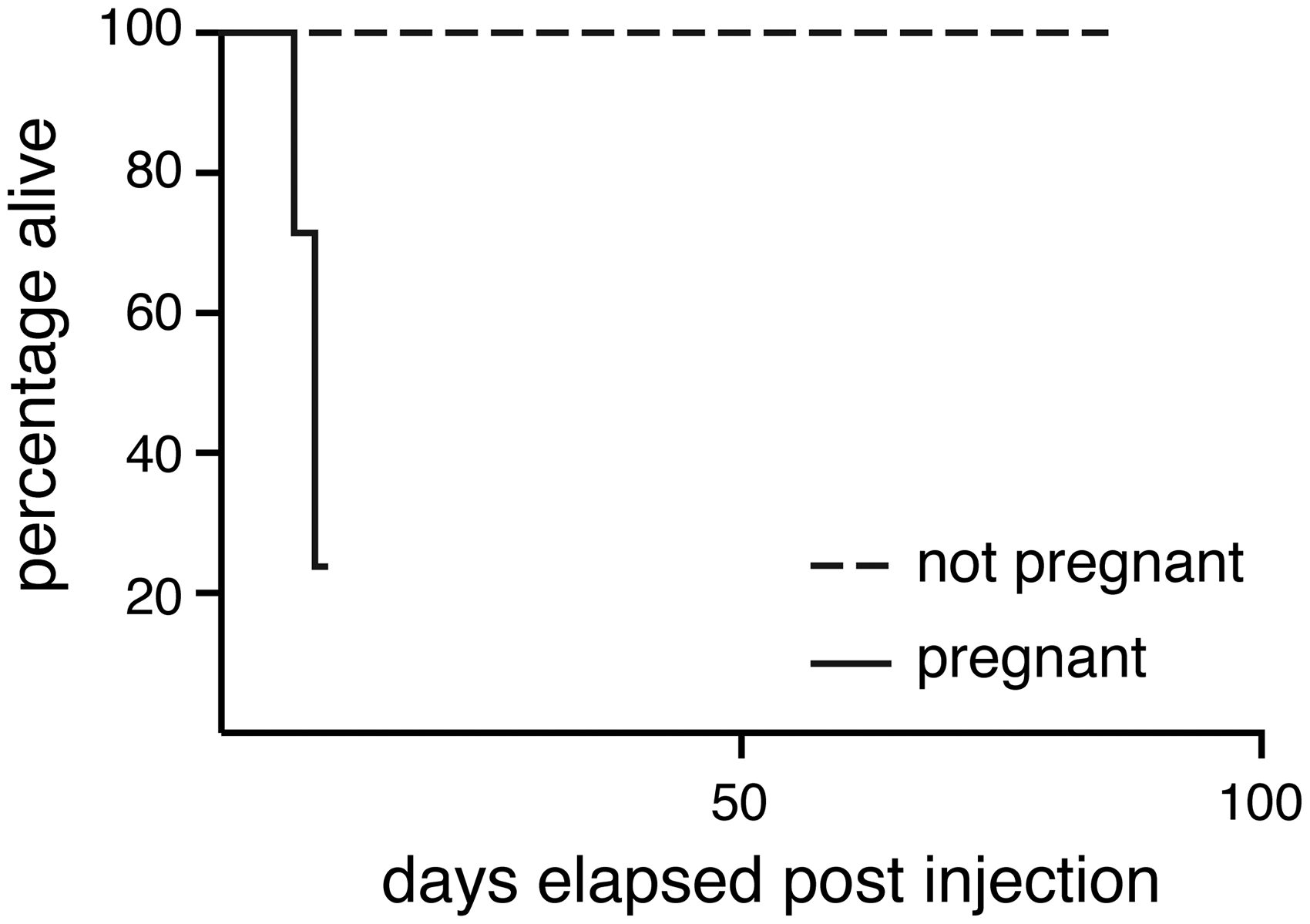
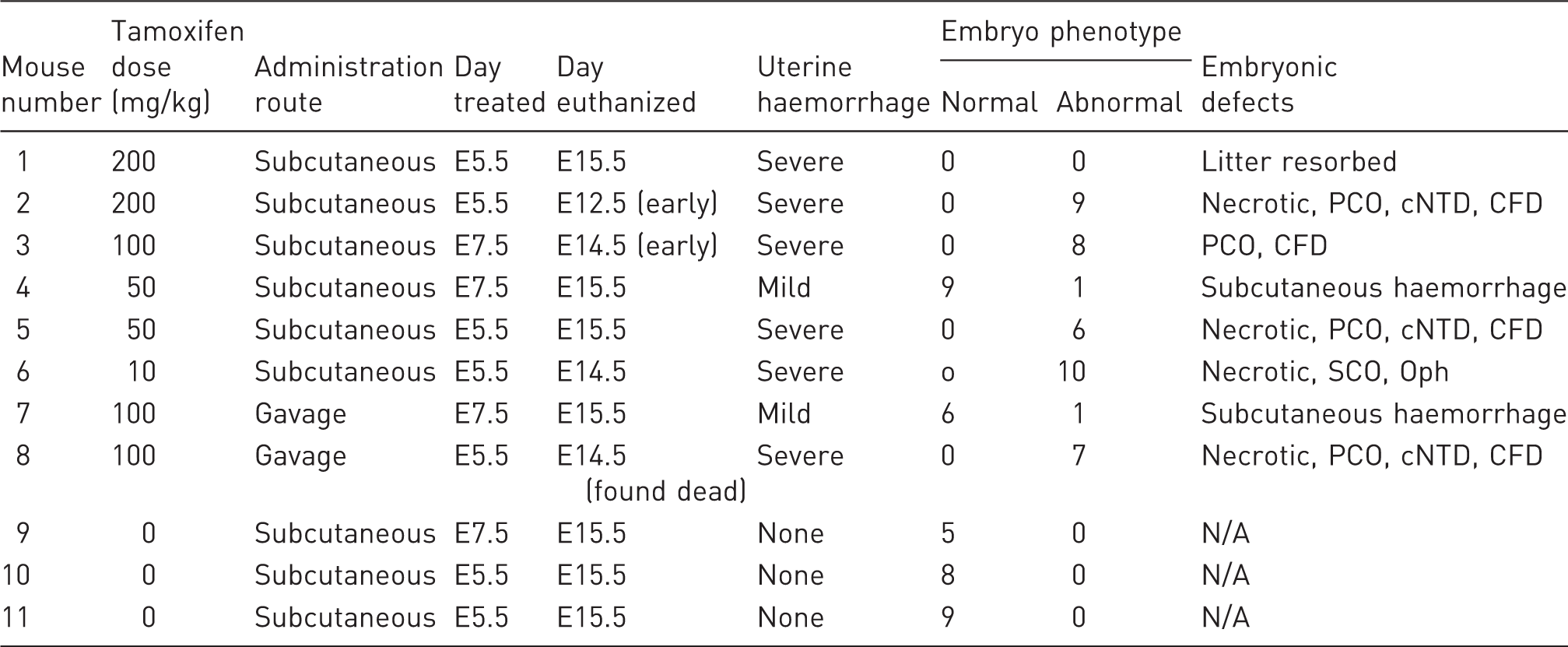
An extensive search of the literature for other examples of maternal tamoxifen toxicity found only a single specific reference to intrauterine haemorrhage resulting from tamoxifen administration. 2 In this case it occurred in 4/5 mice gavaged with 120 mg/kg on E6.5, haemorrhage was only apparent 12 days post-injection, and there were no apparent effects on the embryos. All other studies focused on reduced litter survival after tamoxifen administration, but not maternal health.3–6 In addition to maternal toxicity, we also found that maternal tamoxifen treatment had a teratogenic effect on embryos. We collected all embryos from the six subcutaneously injected and two gavaged mothers. The majority of these embryos showed abnormal development (42/57) compared with controls from three litters of mothers injected with corn oil alone (0/22, p < 0.0001, two-tailed Fisher’s exact test). A wide range of defects were observed, including pericardial and subcutaneous oedema, craniofacial defects, micro-ophthalmia and necrosis. In addition, one litter was completely resorbed (see Table 1). Interestingly, the two litters from mothers with mild uterine haemorrhage had the lowest rate of abnormal embryos. None of these embryos carried a Cre recombinase allele, and therefore we conclude that the observed embryonic defects were either a direct result of tamoxifen exposure or an indirect consequence of maternal uterine dysfunction.
Clinically, it is well known that tamoxifen therapy for the treatment of early-stage hormone-sensitive breast cancer in humans is contraindicated in pregnancy. AstraZeneca reported congenital anomalies in live births from mothers undergoing tamoxifen therapy during pregnancy, particularly when administered in the first trimester. 7 Tamoxifen administration in vivo during pregnancy has been linked to impaired placental development and vascular dysfunction resulting in increased embryonic anomalies and embryonic death in rat. 8 In summary, we believe that although tamoxifen administration during mouse pregnancy is a useful tool for precision alteration of gene expression in the study of mouse embryonic development, it can be harmful to the pregnant mouse if administered too early in gestation. Therefore, we suggest that to comply with the principles of the three Rs, 9 future studies using tamoxifen administration to female mice early in pregnancy should proceed with caution.
Footnotes
Acknowledgements
All animal experiments were compliant with the UK Animals (Scientific Procedures) Act 1986 and approved by the Home Office, UK. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author contribution
DBS and FMA conceived the project. NV and DBS designed the experimental approach. NV, AC and DBS performed experiments. NV and DBS analysed data. NV and DBS wrote the manuscript with support from the other authors. DBS guided the project and obtained funding.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the British Heart Foundation (grant numbers RE/13/1/30181 and FS/17/55/33100). NV is supported by a Novo Nordisk postdoctoral fellowship run in partnership with the University of Oxford.