
Abstract
Traditional serological enzyme-linked immunosorbent assay (ELISA) is routinely used to monitor pathogens during quarantine in most animal facilities to prevent possible infection. However, the ELISA platform is a single-target assay, and screening all targeted pathogens is time-consuming and laborious. In this study, to increase sensitivity and to reduce diagnosis time for high-throughput processes, multiplex PCR and DNA biochip techniques were combined to develop a multi-pathogen diagnostic method for use instead of routine ELISA. Eight primer sets were designed for multiplex PCR to detect genes from seven targeted bacterial and viral pathogens. DNA–DNA hybridization was conducted on a biochip following the multiple PCR analysis. Using this method, a total of 24 clinical samples were tested, and the result showed that not only single infection but also co-infection by multi-pathogens can be detected. In conclusion, multiplex PCR coupled with a DNA biochip is an efficient method for detecting multi-pathogens in a reaction. This platform is a useful tool for quarantine services and disease prevention in animal facilities.
Environmental and genetic factors as well as their interactions have a major impact on laboratory animal welfare and variation in experimental results in scientific studies. 1 Pathogen infection or contamination directly reflects the environmental quality of laboratory animal facilities. 2 Therefore, most laboratory rodents are maintained in specific pathogen free (SPF) conditions to enhance biosafety and to eliminate pathogen infection. 3 It is very important to establish a quarantine program for animal health monitoring in an animal vivarium to ensure the quality of laboratory animals. 2 In most quarantine programs, serological methods are used for screening pathogens that include viruses, bacteria and other organisms. Current serological methods include enzyme-linked immunosorbent assay (ELISA), indirect immunofluorescence assay (IFA), hemagglutination inhibition (HAI) and polymerase chain reaction (PCR). 2 However, ELISA, IFA and HAI methods are laborious and time-consuming, and may have low sensitivity due to the low affinity of anti-pathogen antibodies. 4
PCR and related techniques have become key procedures in pathogen diagnosis.5,6 Since 1993, single-target PCR has been used for specific pathogen detection from blood samples,6,7 for example, Mycoplasma gallisepticum, 4 Mycoplasma synoviae, 8 and reovirus. 9 The limitation of single-target PCR is that it only detects one specific pathogen in each reaction. However, this limitation can be eliminated by using multiplex PCR which includes multi-pair primer sets to amplify molecular targets from different pathogens in one reaction.10,11 The advantages of multiplex PCR include time efficiency, cost effectiveness, and high sensitivity for use in screening and monitoring pathogens in animals.12,13 In most cases, agarose gel electrophoresis is typically used to evaluate the results of multiplex PCR. However, this method is restricted by the limitation in amplicons that can be resolved on a gel. 13
DNA–DNA hybridization biochip technology has become a powerful molecular tool in gene expression studies, 14 ecological research, 15 phylogenetic analysis, 16 and pathogen detection.17,18 The main concept of this technology is that target DNA can specifically and sensitively hybridize to sequence-specific probes on a DNA biochip. This technique can also be coupled with PCR to increase diagnosis sensitivity. 19 Therefore, a DNA–DNA hybridization biochip combined with multiplex PCR has been developed for detection of pathogens, including Salmonella typhimurium, 20 Yersinia enterocolitica 21 and herpesviruses. 22
In this study, a complementary method combining the advantages of multiplex PCR and DNA–DNA hybridization in a biochip system was developed for pathogen detection and quarantine services in SPF animal facilities. Biotinylated multiplex PCR products of target pathogens were captured on immobilized DNA oligonucleotide probes by DNA–DNA hybridization on a DNA biochip, and the hybridized signals were detected by chemiluminescence with a streptavidin–alkaline phosphatase (Strep–AP) conjugate. The results demonstrated that multiplex PCR plus a DNA biochip is an efficient method for the detection and surveillance of seven significant pathogens in laboratory rodents.
Materials and methods
Pathogens and materials
Oligonucleotide primer sequences and amplicon length for each target gene in different pathogens.
F: forward primer; R: reverse primer.
Primer design and synthesis
The PCR primer pairs of specific pathogens were designed using Primer 3 software 24 based on the DNA sequences published in GenBank. Specific primer sets were used to amplify target genes in single-target and multiplex PCR and were selected according to the following criteria: (1) melting point between 55 and 57℃; (2) compatibility with the PCR mixture; (3) amplicons of 100–500 bp; (4) recognition of a single species of pathogens but a different strain of pathogens; and (5) specific analysis by comparison with known gene sequences using the BLAST search program provided by the National Center for Biotechnology Information (NCBI; Bethesda, MD, USA). The primer sequences and the corresponding genes of specific pathogens are also listed in Table 1. All oligonucleotide primer sets were synthesized and 5'-end biotin-labeled by Bio Basic Canada Incorporation (Markham, Ontario, Canada).
Nucleic acid extraction
DNA of pathogens or clinical samples was extracted using the taco Nucleic Acid Automatic Extraction System with a taco DNA/RNA Extraction Kit. The protocols for all samples examined in this study were in accordance with the manual provided by the manufacturers.25,26 To prepare the sample solution, each sample (50 µL) was mixed with 450 µL of phosphate buffer saline in a 1.5 mL microcentrifuge tube, and then centrifuged at 12000
Single-target PCR and multiplex PCR
For the single-target PCR, 10–100 ng of purified DNA was used for each reaction. Single-target PCR amplifications were carried out with 0.25 mM dNTP, 1.5 mM MgCl2, 1 × PCR buffer, 1 U Taq polymerase and 0.5 µM single pathogen primer set in a final volume of 50 µL. Amplification was performed in a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA, USA) under the following conditions: 1 cycle at 94℃ for 5 min, 35 cycles at 94℃ for 30 s, 55℃ for 30 s, and 72℃ for 30 s, and the cycling was ended with a final elongation step for 5 min at 72℃. Amplicons were analyzed using 2% agarose gel electrophoresis in 1 × TAE buffer (40 mM Tris-acetate [pH 8.0], 2 mM EDTA), stained with ethidium bromide (0.5 ug/mL) for UV analysis, and digitized by a Doc Print System (Vilber Lourmat, Marne-la-Vallée, France). Each pathogen specific target fragment was cloned into the plasmid pGEM-T (Promega, Madison, WI, USA), and sequenced by an ABI 3730xl DNA Analyzer (Applied Biosystems) and confirmed by online BLASTN programs (NCBI). Multiplex PCR was performed by mixing all eight primer pairs similarly to the single-target PCR, with adjusted annealing temperature of 54℃ for 30 s, for 30 cycles, and with the cycles ending with a final elongation step for 5 min at 72℃. In order to test the sensitivity of the corresponding single-target PCR and the multiplex PCR, PCR sensitivity experiments were evaluated comparatively in 10-fold serial dilutions of plasmid.
DNA–DNA hybridization and signal generation on DNA biochips
Purified and denatured single-target PCR fragment of a single pathogen was prepared for the DNA biochip probe. DNA biochip probes were spotted onto DNA biochips using the Fast Spot system according to the manufacturer’s manual. The position of each pathogen on the DNA biochip is shown in Figure 4A. DNA–DNA hybridizations were conducted on a DNA biochip following multiplex PCR amplification. First, the biotin-labeled multiplex PCR products were denatured at 95℃ for 5 min and chilled on ice for 2 min. For DNA–DNA hybridization, 5–10 µL of multiplex PCR product was mixed with the hybridization buffer (150 mM NaCl), then added to the reaction chamber and sealed with a lid to prevent evaporation of the mixture. The sealed DNA biochip was incubated in a chip hybridization oven with shaking for 25 min. After DNA hybridization, the biotin signal generation kit was used to generate the signal. In brief, the DNA biochips were washed with washing buffer and the chamber was blocked with blocking buffer for 15 min to avoid non-specific binding. For each chip, 0.2 µL of Strep–AP was mixed with 200 µL of blocking buffer and allowed to react for 10 min at room temperature (RT). The DNA biochip was then washed twice with washing buffer, followed by an addition of enzyme substrate, 5-bromo-4-chloro-3-indolyl phosphate, and nitro blue tetrazolium solution. Signal generation was allowed in the dark for 7–15 min. The development of a purple color during incubation indicated the presence of the target DNA.
Results
Single-target PCR
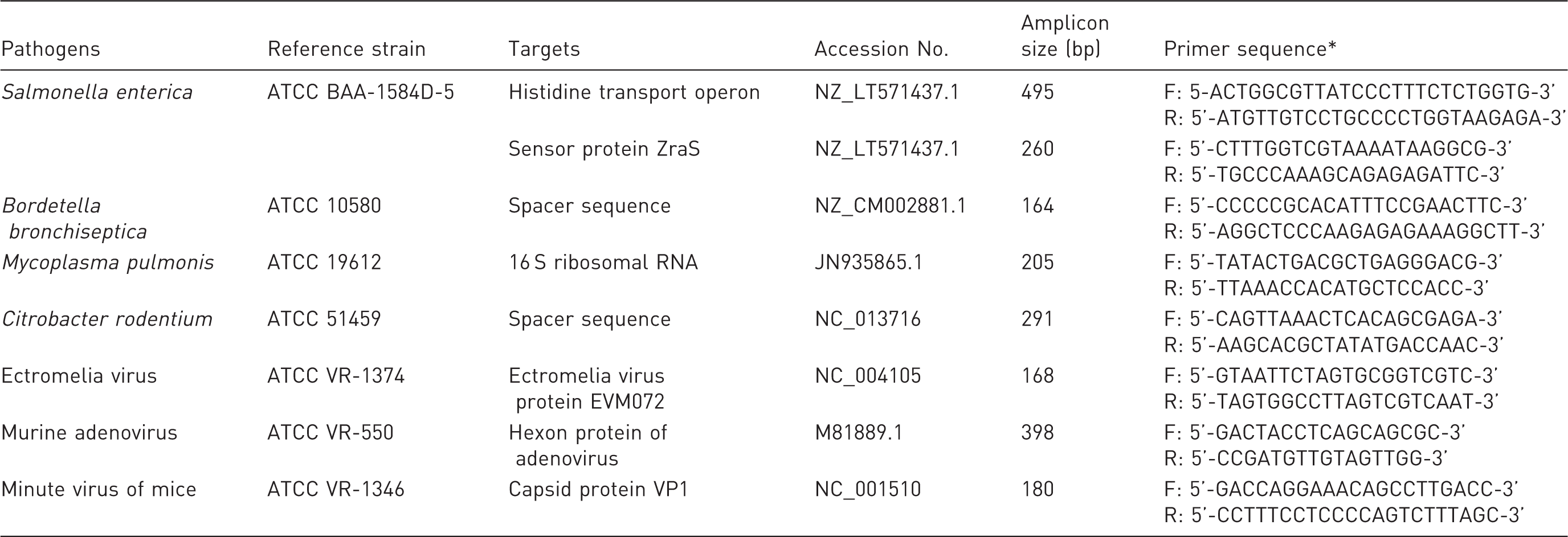
In this study, four bacterial and three viral pathogens were detected. Single-target PCR was tested with a corresponding reference template extracted from the reference strain of the pathogen (Table 1). The criteria for primer design were described in the Materials and methods section. Eight primer pairs targeting specific sequences of pathogens and PCR conditions were systematically used for the single-target and multiplex PCR. Specific primer pairs for each pathogen were first analyzed in the single-target PCR which produced amplicons of 495, 260 bp for Salmonella and 164, 205, 291, 168, 398, 180 bp for Bordetella, Mycoplasma, Citrobacter, ectromelia virus, murine adenovirus and minute virus of mice, respectively. Each target gene of the pathogen could be specifically amplified using its defined primer pair, and PCR products showed the expected size ranges as shown in Figure 1. Specific PCR fragments of each pathogen were cloned into the plasmid pGEN-T for DNA sequencing and sensitivity test of the single and multiplex PCR.
Primer specificity test by single-target PCR of pathogen. Lane M is the DNA 100 bp ladder markers with specific base pairs (bp) indicated on the left. Lanes 1 to 8 are the single-target PCR results containing the template from each of the reference pathogen strains, Salmonella (1 and 2), Bordetella (3), Mycoplasma (4), Citrobacter (5), ectromelia virus (6), adenovirus (7) and minute virus (8).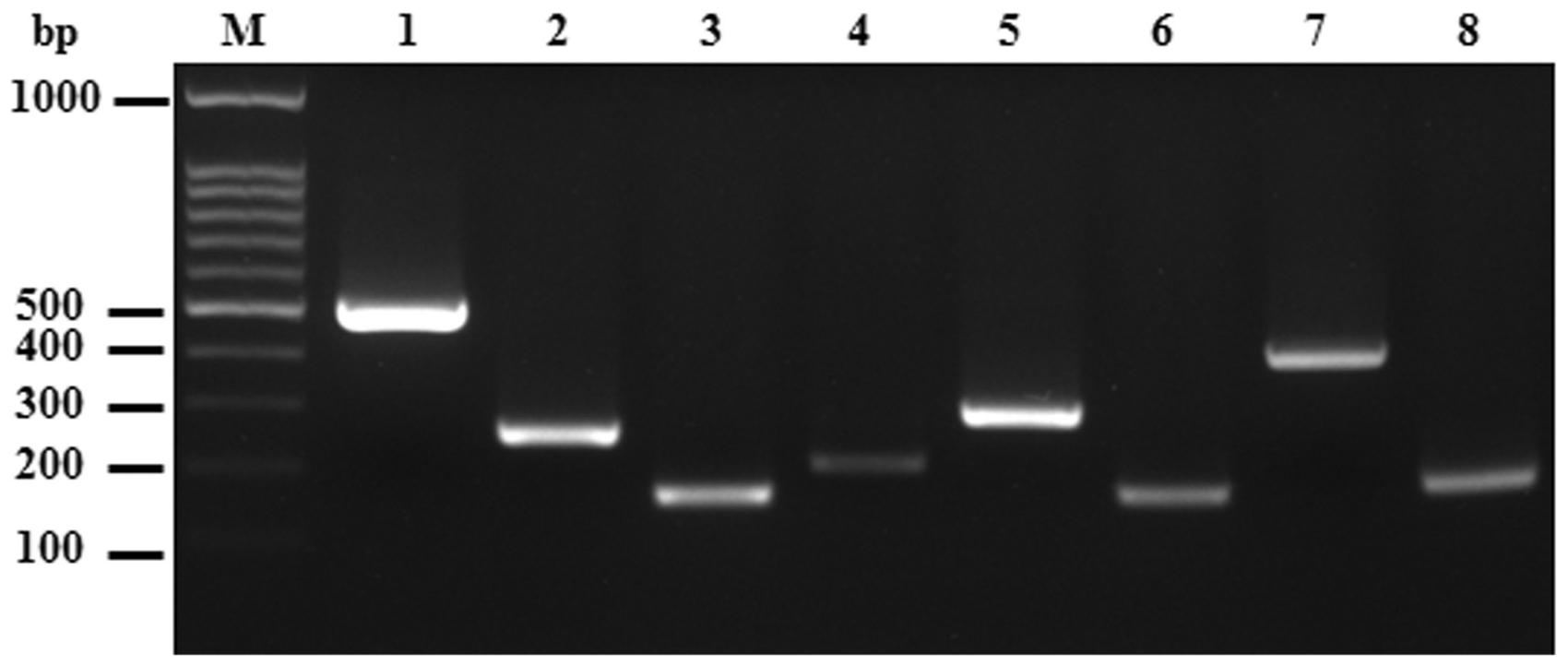
Sensitivity of single and multiplex PCR
After DNA sequences of all amplified PCR fragments were confirmed by the online BLASTN program, the PCR sensitivity experiments were performed in 10-fold serial dilutions of plasmid constructs containing the specific pathogen target fragment. The minimum quantities detected by the single-target PCR were 2.64 × 101, 2.90 × 102, 2.87 × 102, 2.79 × 101, 2.90 × 101, 2.70 × 101 and 2.89 × 102 copies for Salmonella, Bordetella, Mycoplasma, Citrobacter, ectromelia virus, murine adenovirus and minute virus of mice, respectively (Supplementary Figures S1–S7, see http://lan.sagepub.com online for all supplementary material in this article). The detection limits for the multiplex PCR were 2.64 × 101, 2.90 × 103, 2.87 × 102, 2.79 × 102, 2.90 × 102, 2.70 × 102 and 2.89 × 103 copies for Salmonella, Bordetella, Mycoplasma, Citrobacter, ectromelia virus, murine adenovirus and minute virus of mice, respectively (Figure 2). The results indicated that multiplex PCR can be conducted in a one tube reaction by our primer sets, and that the multiple PCR was slightly less sensitive than the single-target PCR.
Sensitivity of multiplex PCR for simultaneous amplification of all pathogen target DNA. Lane M is the DNA 100 bp ladder markers with specific base pairs (bp) indicated. Serial 10-fold dilutions of plasmid constructs which contained the specific pathogen target fragments. PCR product loci of each pathogen were the same as in single-target PCR (Figure 1). *DNA dilution. 100 = 1 ng plasmid DNA. Pathogen test on mouse clinical samples by multiplex PCR. Lane M is a molecular weight 100 bp ladder marker. Lane 1 is the #7 clinical sample that is pathogen positive for Citrobacter (291 bp), ectromelia virus (168 bp) and minute virus (180 bp). Lane 2 is the #15 clinical sample that is pathogen positive for Bordetella (164 bp), Mycoplasma (205 bp) and Salmonella (495 bp and 260 bp). Lane 3 is the #22 clinical sample that is pathogen positive for murine adenovirus (398 bp).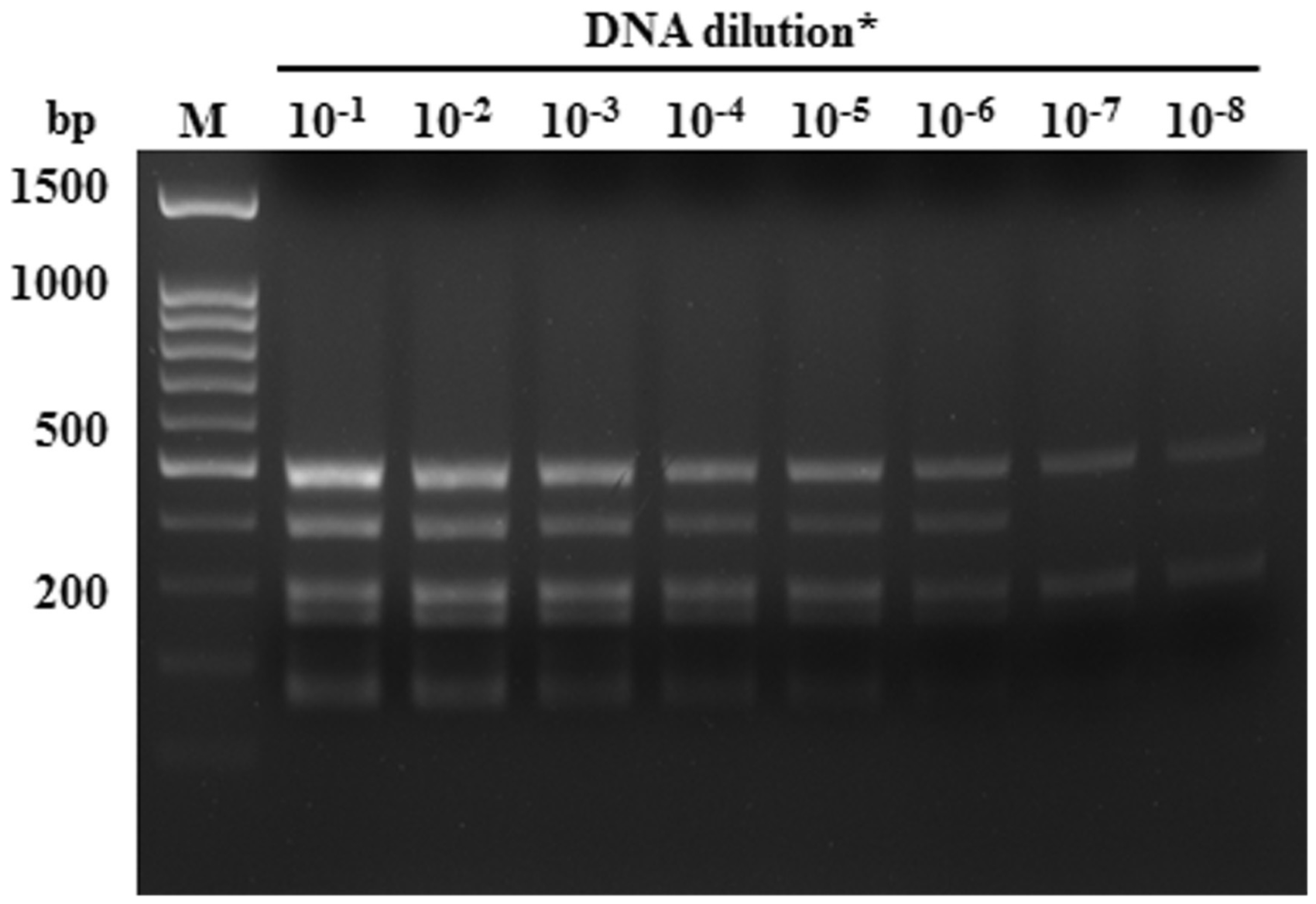
Screening of clinical specimens by multiplex PCR coupled with DNA biochip hybridization
Number of positive samples in 24 mouse clinical blood samples detected for seven pathogens by multiplex PCR DNA biochip and ELISA.

*Test results are presented by number of positive and tested samples. †Total 24 clinical samples were labeled from #1 to #24.
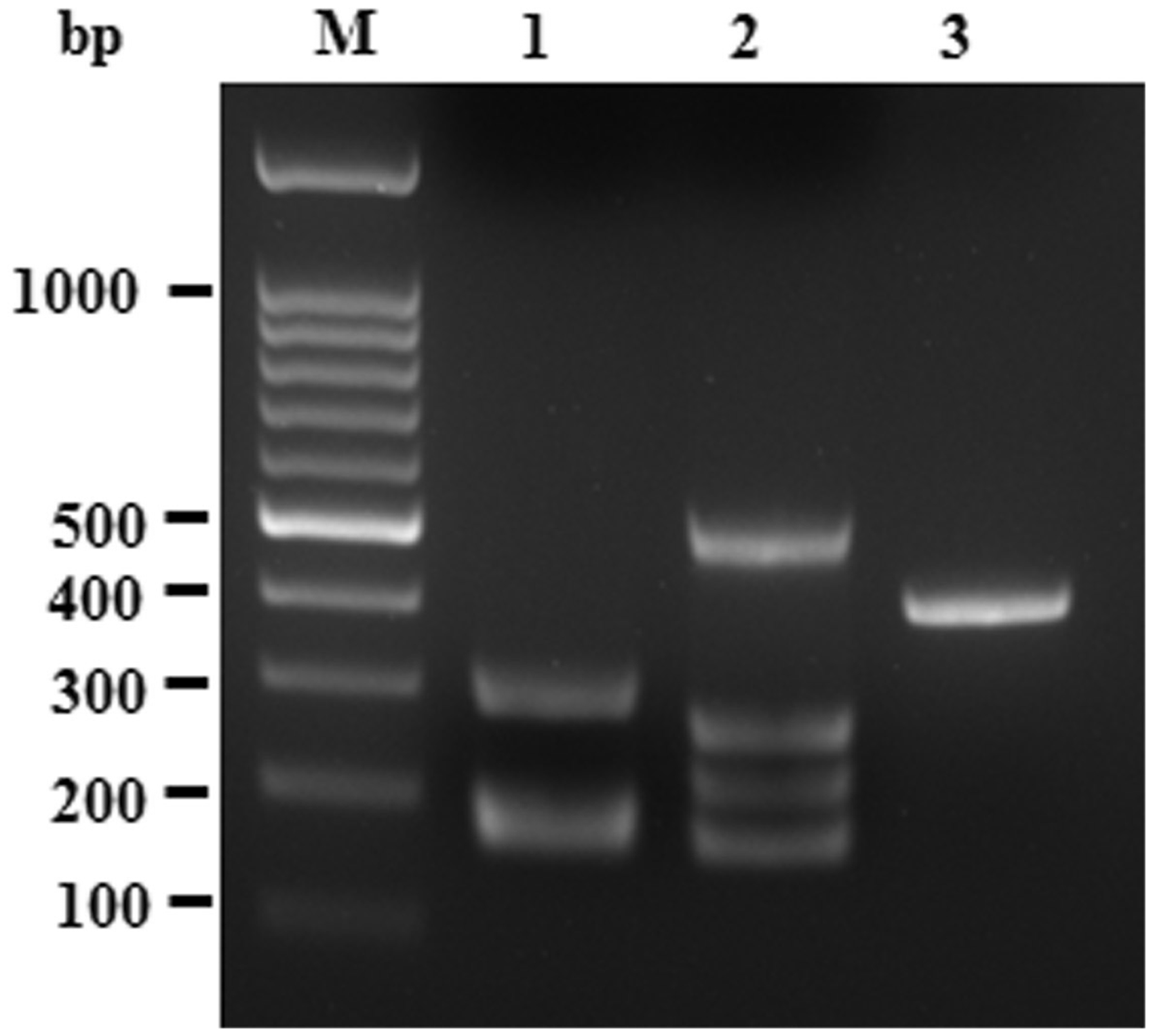
In multiplex PCR, all target genes were amplified with 5'-end biotin-labeled primer to generate biotinylated DNA fragments. Amplified multiplex PCR products were added into the biochip reaction wells following the DNA–DNA hybridization and the signal generation procedure, as described in the Materials and methods section. In this study, the DNA biochip of specific pathogens contained eight different probes that were able to recognize seven different pathogens. The spots of the pathogen positive responses corresponded with the pathogen spot map shown in Figure 4a. DNA samples from the multiplex PCR products shown in Figure 3 were analyzed in DNA biochip hybridization. Figure 4b shows the hybridization result of the multiplex PCR product from the Citrobacter, ectromelia virus and minute virus positive samples. The results showed four spots of Bordetella, Mycoplasma and Salmonella, and one spot of murine adenovirus (Figures 4c and 4d). The results of the DNA–DNA hybridization of DNA biochips were the same as those in multiplex PCR (Figures 3 and 4). Three of the 24 clinical blood samples were positive using ELISA and also positive using multiplex PCR combined with DNA biochip. Samples which were co-infected with multiple pathogens could also be simultaneously detected by multiplex PCR coupled with DNA biochip.
Pathogen detection of multiplex PCR product through DNA–DNA hybridization on a DNA biochip using a biotin streptavidin–alkaline phosphatase (Strep–AP) indicator system. (a) Pathogen spot map on the DNA biochip. Each chip includes nine probes to detect the following genes from different pathogens: Bordetella (1), Mycoplasma (2), Salmonella (3 and 4), Citrobacter (5), ectromelia virus (6), adenovirus (7), minute virus (8) and the hybridization control (C+). (b–d) Results of multiplex PCR products from various pathogen-positive samples hybridized on DNA biochips. Chip B (b) is a hybridization result of the multiplex PCR product from a Citrobacter, ectromelia virus and minute virus positive clinical sample #7. Chip C (c) is a hybridization result of a multiplex PCR product from a Bordetella, Mycoplasma and Salmonella positive clinical sample #15. Chip D (d) is a hybridization result of multiplex PCR product from a murine adenovirus positive clinical sample #22.
Discussion
For surveillance, diagnosis and control of disease, the principal methods for microbial detection in animal populations are serological tests. 27 In most SPF animal vivarium quarantine programs, ELISA and IFA are the most often used serological methods for screening pathogenic viruses, bacteria and other organisms. 2 ELISA and IFA are singleplex assays in which only one antibody reacting with a single pathogen is measured in each reaction. However, in most SPF animal quarantine programs, more than 20 pathogens need to be detected. Therefore these methods may be laborious and time-consuming, with a low sensitivity resulting from a low affinity of the anti-pathogen antibody.2,4 Moreover, immunodeficient mice are now widely used in biomedical and preclinical studies, which are housed in most SPF facilities. 28 However, immunodeficient mice have defects in T- and B-cells and lower concentrations of serum immunoglobulins for ELISA or IFA analysis.29,30 Therefore, the aim of this study was to develop a high-throughput, time-efficient, multiplex and economical method for use in quarantine programs and sentinel animal programs. Here, multiplex PCR and DNA biochip hybridization were combined to provide a diagnostic platform with higher sensitivity and specificity, as well as allowing easier interpretation than traditional serological tests.
Multiplex PCR is a popular platform for the detection of pathogenic bacteria or viruses.10,12,31 The advantage of multiplex PCR is that it uses more than one specific target primer pair with appropriate length, similar annealing temperature and base composition to ensure a multi-target amplification in a single reaction.5,10,11 However, a major drawback of multiplex PCR is that with multiple primer pairs of various pathogens, non-specific amplicons can be formed easily. 32 Therefore, Tm value, 33 G/C content and specificity34,35 of primers are crucial factors for a successful multiplex PCR. In this study, eight primer pairs were designed for seven pathogens based on specific sequence, similar G/C content, appropriate amplicon length and compatibility of multiple primer mixture. In the initial test, the same concentrations of each pathogen primer set were used in the single-target or multiplex PCR. However, there were fewer amplicons of ectromelia virus, murine adenovirus and minute virus than other pathogens by gel electrophoresis (Supplementary Figure S8). A similar situation has also been found in the detection of Vibrio parahaemolyticus strains by multiplex PCR. 36 Therefore, the individual primer concentration was adjusted, resulting in improved multiplex PCR. For example, the primer concentrations of murine adenovirus and minute virus were adjusted to 0.125 µM each in the primer mixture, and this multiple primer mixture was able to detect all pathogens in the multiplex PCR.
Agarose gel electrophoresis is typically used to evaluate multiplex PCR results. However, this method is limited by the number of samples that can be resolved on one gel. 13 It is also difficult to distinguish amplicons with similar length, for instance amplicons of ectromelia virus (168 bp) and minute virus (180 bp) (Figure 3, lane 1). In order to improve the results and expedite analysis, the DNA biochip hybridization technique was used to replace gel electrophoresis after multiplex PCR. Compared with gel electrophoresis, DNA biochip can increase the specificity and reliability by decreasing the occurrence of false-negative and false-positive results through hybridization processes. 37 With DNA biochips, gene-specific oligonucleotide probes, either fluorescent- or radioactive-labeled probes, and a scanner represent the common method of detection; however, the need for sophisticated equipment and dangerous radiation are disadvantages which limit its broad application in standard laboratories.37–39 Here, single-target PCR products of each pathogen were used as probes on DNA biochips and Strep-AP-conjugated color visual DNA biochip format was used to provide a more convenient and economical method for examining hybridization efficiency and accuracy of biotin-labeled multiplex PCR products (Figure 4). In previous studies, NaCl concentration of the hybridization buffer 40,41 and the hybridization temperature 42 have been reported to be the main factors affecting the efficiency of DNA hybridization. In this study, different temperatures and NaCl concentrations ranging from 40℃ to 55℃ and 50 to 200 mM were used to test hybridization efficiency. The optimal conditions were determined to be 50℃ and 150 mM of NaCl for DNA–DNA hybridization, and were subsequently used in the hybridization step of the DNA biochip.
Currently, not only multiplex-PCR but also probe-based quantitative PCR (qPCR) are widely used for the detection of pathogens in rodents. These two PCR-based methods have similar advantages, i.e. high sensitivity, reproducibility, and with only small amounts of clinical samples being needed. However, most probe-based qPCR involving fluorescent labeling has at least one of the following disadvantages: (1) expensive labeling of staple strands, (2) fluorophores bleach, (3) unsuitability for large-scale multi-fluorescent labeling, and/or (4) sophisticated and expensive equipment.43,44 In this study, we developed a multiplex PCR plus DNA biochip method for rodent pathogen detection, which requires no fluorescent labeling, and with all the necessary equipment being the same as that for the conventional PCR technique.
In conclusion, multiplex PCR coupled with DNA biochip hybridization provides a diagnostic format which is sensitive, specific, accurate, and more easily-interpreted than conventional single-target PCR, qPCR, IFA and ELISA. Therefore, the multiplex PCR coupled with DNA biochip platform is a very useful tool for pathogen detection and disease prevention, and for use by animal quarantine services in SPF level animal facilities.
Footnotes
Acknowledgements
The authors thank the Laboratory Animal Core Facility funded by the Agricultural Biotechnology Research Center (ABRC) at Academia Sinica for technical support and pathogen tests of clinical samples. The authors are also grateful to Ms Miranda Loney of the Academia Sinica for her critical reading of this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this research was funded by The Ministry of Science and Technology in Taiwan, ROC (Reference: NSC 101-2324-B-001-001).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.