
Abstract
In addition to carbon tetrachloride (CCl4), thioacetamide (TAA) represents a second widely used model for the induction of experimental liver fibrosis, but can also be employed for the development of acute liver failure and liver tumours. While TAA itself is not hepatotoxic, its reactive metabolites covalently bind to proteins and lipids thereby causing oxidative stress and centrilobular necrosis. Compared with CCl4, TAA leads to more periportal infiltrates and more pronounced ductal proliferation. While TAA has been shown to induce liver fibrosis development in several different mouse strains, wide variations in the administration routes, doses and treatment durations have been reported. Therefore, an adoption of a universal standard operating procedure for the administration of TAA is urgently needed. For that purpose, we are presenting here two TAA models (intraperitoneal administration of 150 mg/kg of TAA three times per week for 11 weeks in rats, and TAA administration in drinking water at 300 mg/L for 2–4 months in mice) with which we have had success in reliably and reproducibly developing chronic liver injury and fibrosis.
Historical background and current usage
Usage of thioacetamide-induced liver damage in rodents.
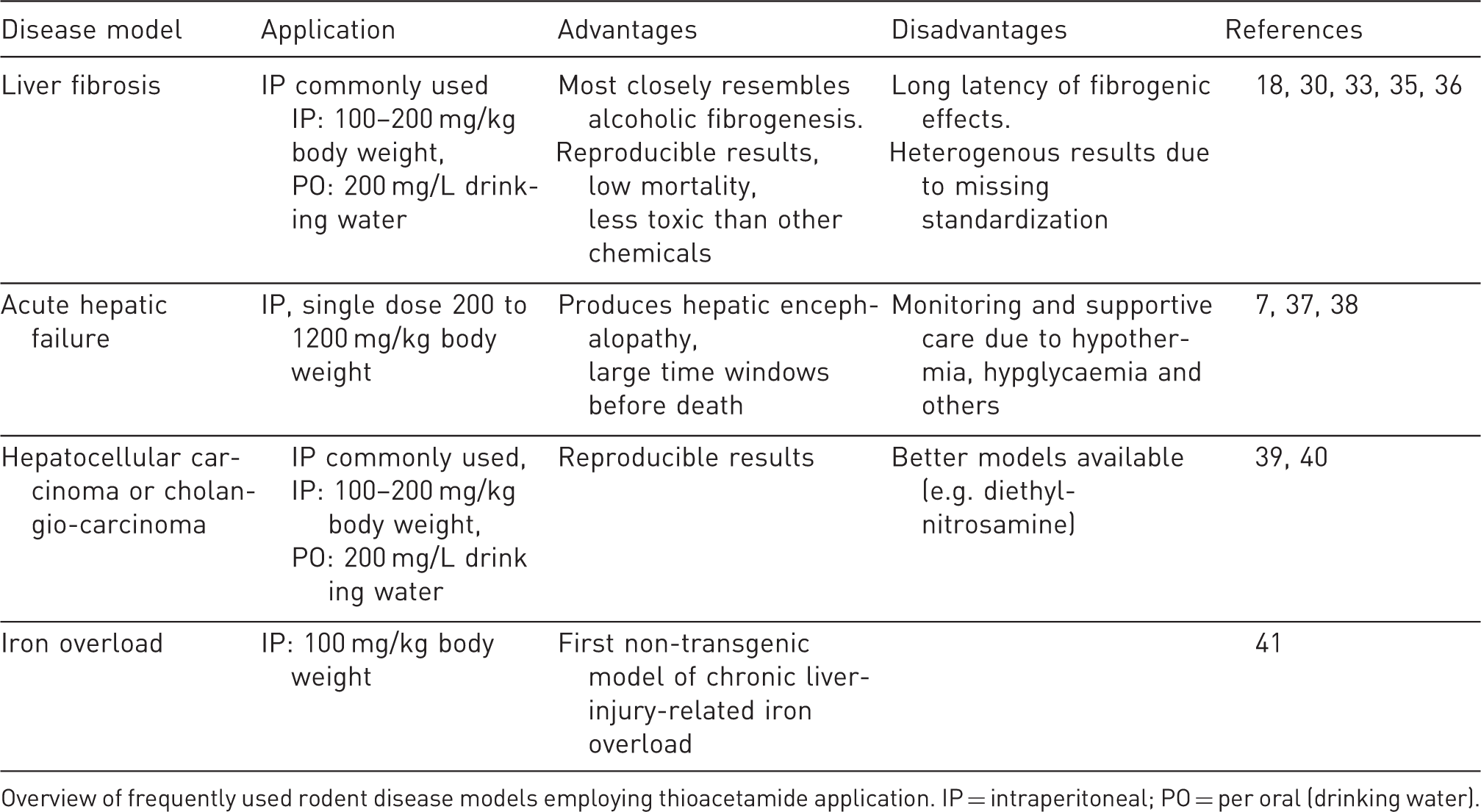
Overview of frequently used rodent disease models employing thioacetamide application. IP = intraperitoneal; PO = per oral (drinking water).
The use of chronic TAA varies widely, particularly in the route of administration; dosage, timing and length of treatment; and animal used (Table 2). In the first half of 2014, there have been 18 papers published in peer-reviewed journals that utilized chronic TAA to produce fibrosis or cirrhosis experimentally (references available upon request). Thirteen studies used rats of various genetic backgrounds (Sprague–Dawley, Wistar, F344, and albino) and five used mice of either C57BL/6 or Kunming backgrounds. The majority delivered TAA by the IP route (12); the remainder (5) used the oral route, three of which used a fixed dose (300 mg/L in drinking water), the others (2) a variable, weight-based dose. One paper did not report the route of administration. The oral and IP routes were used in both mice and rats. Dosages for the IP route ranged from 10 mg/kg to 400 mg/kg, two to three times per week from between 5 and 20 weeks while recently most researches have used dosages of between 100 mg/kg and 200 mg/kg, three times per week for at least six weeks. Most used TAA as the only model of fibrosis and cirrhosis (12); the remainder (6) used TAA in conjunction with other models such as CCl4 or bile duct ligation, for example. Unfortunately, neither the proportion of experimental animals which developed advanced fibrosis or cirrhosis, nor the mortality rates were reported in these studies. It is therefore difficult to make recommendations regarding the most appropriate regimen based on recently published studies. Local expertise and personal experience with the model is likely to be an important factor when planning an experiment with TAA. Here we present our experience with the TAA model, which includes chronic oral exposure in C57BL/6 mice at a fixed dose (300 mg/L in drinking water) for 2–4 months,
13
IP injection of TAA in Sprague–Dawley rats for 11 weeks with or without an experimental drug,
14
and a time-course pilot study of TAA by IP injection in Sprague–Dawley rats for 12 weeks (Figure 1). We report the rate and degree of fibrosis and cirrhosis development, changes in portal pressures and liver collagen content, as well as the mortality rate in the experimental animals.
Time course experiment of thioacetamide (TAA) by intraperitoneal (IP) injection and phosphate-buffered saline (PBS) controls in rats. (a) Aspartate aminotransferase (AST) (upper panel) and alanine aminotransferase (ALT) (lower panel) levels spiked initially and then returned to normal at week 4. (b) Several fibrosis related genes demonstrated consistent elevation throughout the course of the experiment. (c, d) Portal pressures (c) and collagen content in liver tissue (d) plateaued at week 8. Col1A1: collagen type 1α1; Asma: α-smooth muscle actin; Pdgrfb: Platelet-derived growth factor receptor type β; Mmp2: matrix metalloproteinase-2; Timp2: tissue inhibitor of matrix metalloproteinases 2; GAPDH: glyceraldehyde phosphate dehydrogenase. Representative examples of thioacetamide usage in mouse models of experimental liver fibrosis. The main application routes for thioacetamide treatment are intraperitoneal injection or administration in drinking water. IP = intraperitoneal; PO = per oral (drinking water), wk = week.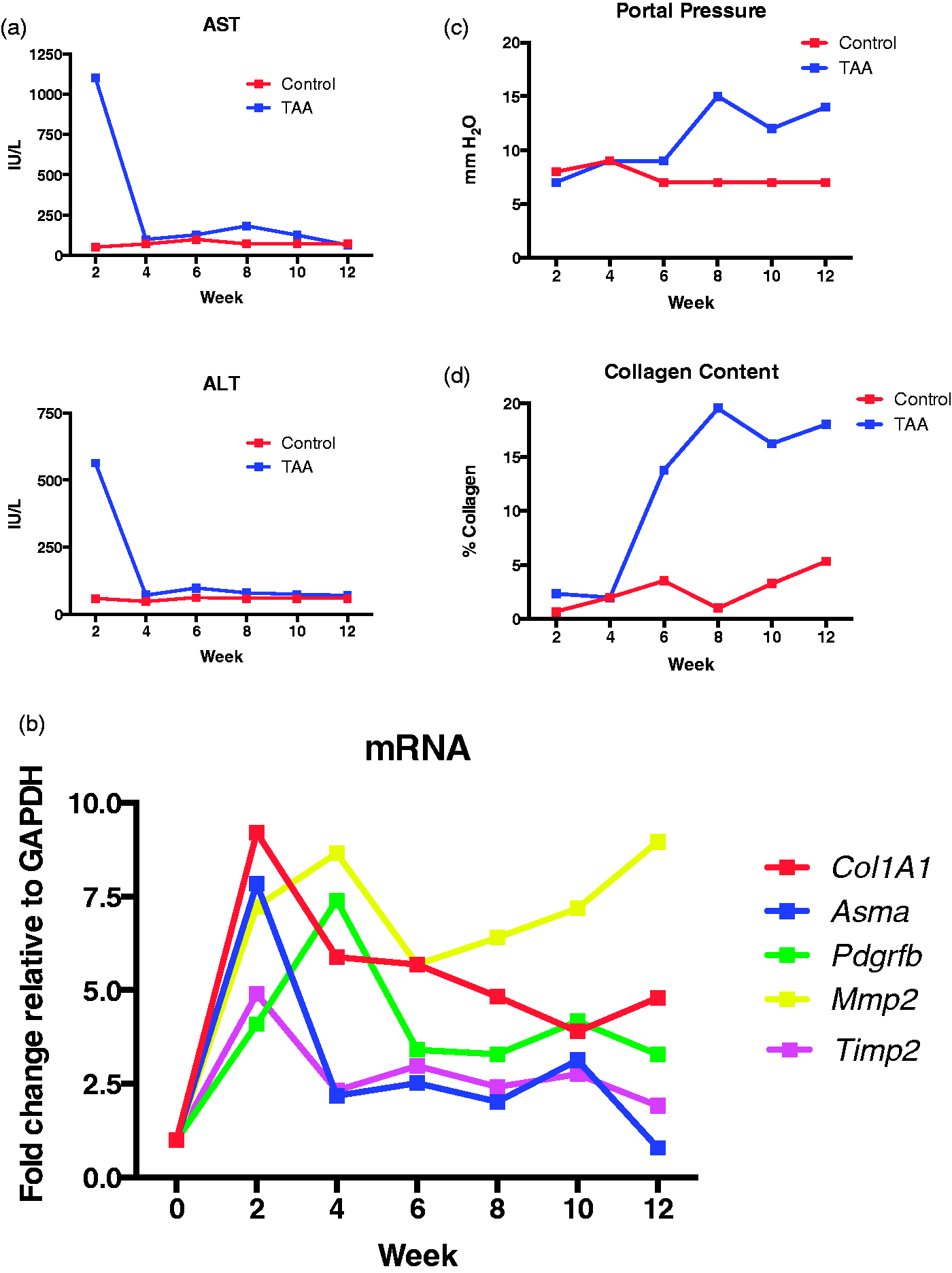

Mechanisms and pattern of liver injury
Despite the hepatotoxic properties of TAA being well known since 1948, the mechanisms by which it causes liver injury are complex and remain poorly understood. TAA requires oxidative bioactivation of its toxic metabolites to cause liver injury that affects both hepatocytes and cholangiocytes.15,16 With regard to the latter, an enlargement of portal tracts without signs of cholestasis is frequently observed.17–19 TAA is first oxidized to TAA-S-oxide and then to TAA-S,S-oxide (Figure 2); this process can be carried out by hepatic cytochrome P450 enzymes or FAD-containing monooxygenases.
20
The lethal dose (LD50) in rats and mice after both oral and IP administration was determined to be approximately 300 mg/kg body weight.
21
Even at high doses, TAA is not toxic to cultured hepatocytes while TAA-S-oxide and TAA-S,S-oxide cause toxicity as demonstrated by increases in cytosolic lactate dehydrogenase (LDH) release.
20
The oxidation of TAA appears to be mediated primarily through CYP2E1, although under varying physiological conditions, the FAD-containing monooxygenases may play a more important role.20,22,23 When Cyp2e1-null mice were acutely exposed to TAA, they were protected from liver injury, elevation in markers of inflammation, and oxidative stress. By contrast, wild-type mice showed markedly increased mRNA levels of tumour necrosis factor (TNF)-α, interleukin (IL)-6 and glutathione peroxidase plus developed centrilobular necrosis and inflammation.
22
In addition to causing oxidative stress, glutathione depletion and thus cellular injury, the reactive metabolites of TAA covalently bind to cellular proteins and lipids. Moreover, this results in alterations in cell cycle-related proteins, protein folding, enzyme inhibition and mitochondrial/chaperone function, all of which leads to significant cytotoxicity.24–26
Simplified model for the formation of noxious thioacetamide (TAA) metabolites and their effects in hepatocytes. In a first biotransformation step, TAA is reversibly metabolized to TAASO. In a second oxidation step, the highly reactive species TAASO2 is formed. Subsequently, TAASO2 can directly react with amine groups (R-NH2) on cellular proteins or lipids resulting in dysfunction. In addition, TAA gives rise to glutathione depletion and elevated levels of reactive oxygen species (ROS), lipid peroxides, cytotoxicity and mitochondrial injury. Together the events may lead to apoptosis, necrosis and the formation of hepatocellular carcinoma and cholangiocarcinoma. Details of the sequence of chemical reactions induced by TAA can be found elsewhere.20,34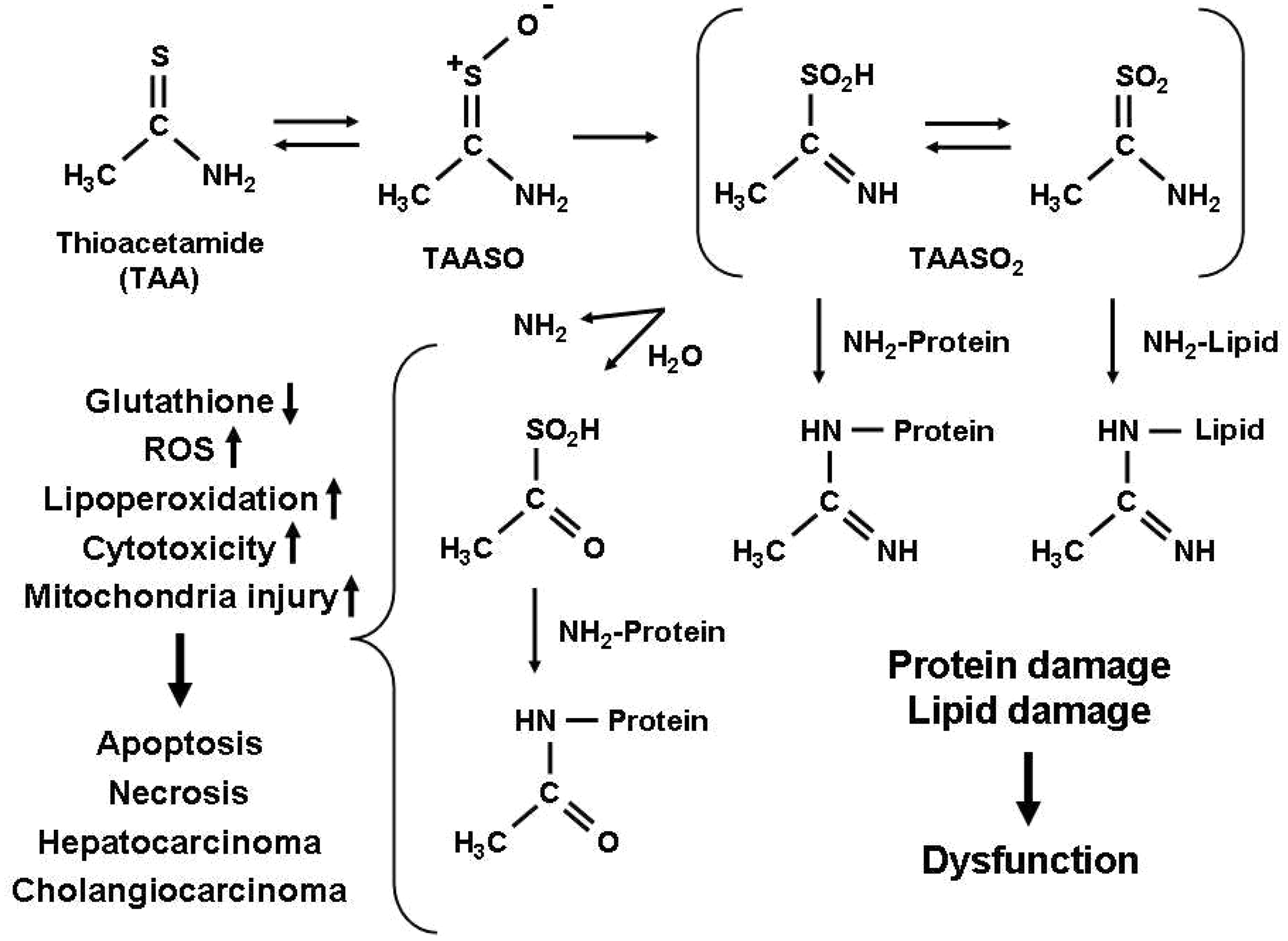
The administration of TAA causes a predictable and reliable dose- and time-dependent pattern of liver injury that resembles human fibrosis and cirrhosis.10,27 With an increased length of intoxication, rodent liver microscopically demonstrates more clearly a mixed inflammatory infiltrate of the portal tract with bile duct proliferation and some hepatocyte necrosis, than a typical pattern of fibrosis with portal–portal and portal–central septa progressing to eventual features of macronodular cirrhosis with bridging fibrosis and regenerative nodules.
Although both CCl4 and TAA result in centrilobular liver damage, several differences between the models should be noted.18,28 TAA displays a longer latency between the exposure to the drug and the development of liver injury.3,29 TAA also leads to more periportal infiltrates and more pronounced ductal proliferation, appearance of acidophilic bodies, and vacuolated nuclei with prominent nucleoli, as well as more pronounced hepatocyte vacuolization.18,19 Bile duct ligation and repeated application of CCl4 have traditionally been employed as the most widely used experimental model of fibrosis. Common to all these three models is the development of hepatic fibrosis that can be most easily visualized with Sirius Red staining (Figure 3).
Histological characterization of the most widely used experimental liver fibrosis models. To visualize the most widely used experimental models, liver sections were prepared from untreated animals, mice subjected to bile duct ligation (BDL) for three weeks or administered carbon tetrachloride (CCl4) (2×/week, 0.2 mL/kg for 12 weeks) and thioacetamide (TAA) (3×/week 100 mg/kg for 15 weeks), respectively. Haematoxylin and eosin (H&E) staining was employed for the overall histological evaluation whereas Sirius Red staining was carried out to evaluate the extent of liver fibrosis. Scale bar: 100 µm (top), 200 µm (bottom).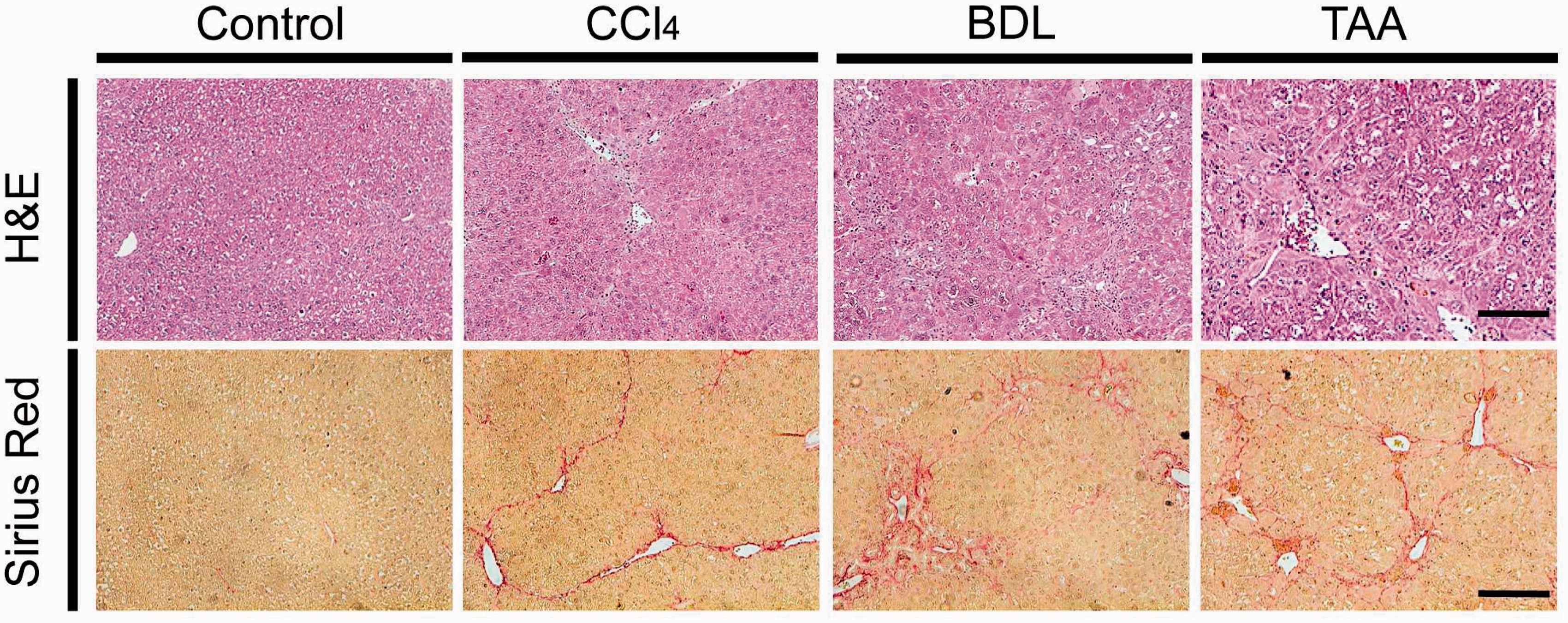
With prolonged treatment using TAA (18 weeks and longer), cellular atypia may become evident and mitotic figures may be observed. 10 As demonstrated in the original TAA publication, 2 chronic intoxication frequently results in malignancy, although TAA is currently rarely used in isolation as a model of HCC or cholangiocarcinoma. In an oral, weight-based TAA model, rats developed portal hypertension and splenomegaly after 12 weeks of exposure, indicating a strong functional relationship to human cirrhosis. 10 Additionally, and perhaps most importantly, rodent hepatic stellate cell activation in TAA intoxication is central to the observed fibrosis and cirrhosis development. 30
To validate the model, we performed a time-course experiment in which Sprague–Dawley rats were exposed to TAA by IP injection (150 mg/kg, three times/week) for 12 weeks. One group (2 experimental animals and one control phosphate-buffered saline [PBS]-injected animal) was sacrificed every 12 weeks and assessment was performed of various fibrosis associated genes by quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR), portal pressures by portal vein cannulation, biochemical analysis of liver injury by serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) measurements, and quantification of collagen content by the BIO-QUANT Life Science morphometry system® (BIOQUANT Image Analysis Corp, Nashville, TN, USA). The results are presented in Figure 1. After an initial spike in inflammation and liver injury at two weeks as assessed by serum ALT and AST, these values returned to normal at week 4. Sustained mRNA elevation of fibrosis associated genes including collagen 1A1 (Col1A1), α-smooth muscle actin (Asma), platelet-derived growth factor receptor β (Pdgfrb), matrix metalloproteinase 2 (Mmp2) and tissue inhibitor of metalloproteinase 2 (Timp2) were demonstrated. Both portal pressures and collagen content of liver tissue reached maximal levels in TAA rats at week 8 and then plateaued. These results reinforce the efficacy of the TAA IP injection strategy for the production of significant fibrosis between 8 and 12 weeks.
General considerations
Safety concerns: Based on the high toxicity of TAA, personal protective equipment including eyeglasses, protective gloves, appropriate clothing and respiratory mask are necessary whenever handling this hepatotoxin. To avoid chemical decomposition, TAA should be stored in a cool, dry and safe place to which only authorized staff have access (e.g. poison cabinet). Genetic background: While the susceptibility to liver fibrosis development in different experimental models has been shown to display significant strain differences,
31
no systematic analyses on strain-specific susceptibility to TAA have been reported. In fact, the successful induction of TAA-mediated liver fibrosis has been described in a variety of genetic backgrounds of both mice and rats.18,32 Route, dosage, timing and duration of treatment: As discussed above in ‘Historic background and current usage’, there is wide variation in currently used TAA models and it is beyond the scope of this standard operating procedure (SOP) to discuss the differences. As a guide to planning a TAA experiment, we present here two models with which we have both personal experience and confidence in their safety and efficacy for fibrosis and cirrhosis induction. Experimental uses and design: A typical TAA experiment involves the comparison of the rate or degree of fibrosis or cirrhosis development between a control group and a treatment or intervention group. In this era of rapidly increasing interest in anti-fibrotic therapies, it is a convenient and reliable tool for the relatively rapid in vivo screening of potential therapies, as it allows for the generation of results within 4–5 months. The timing of the introduction of the experimental therapies can be modified to assess their effectiveness in modulating different phases of fibrosis. The TAA model may also be used to study functional consequences of cirrhosis such as portal hypertension, and as previously mentioned, acute TAA exposure in high doses can be used to study acute liver failure and encephalopathy. Finally, TAA administration may also be used to study the biology of fibrosis, as it provides a reliable stimulus for stellate cell activation and fibrosis development. Genetic knockdown and overexpression techniques are best utilized in this way. The remainder of this SOP will focus on the use of TAA to produce fibrosis and cirrhosis. Analysis of treatment and control cohorts: Selection of readouts will depend on the aims of the project. In a study in which the degree of fibrosis development is the major outcome being assessed, a thorough analysis of liver histology plus a fibrosis quantification technique should be used. Techniques used will depend upon local expertise, but important considerations are the use of an experienced liver histopathologist to assess random sections in a blinded fashion. We suggest formalin-fixed, paraffin embedded liver sections should be stained with haematoxylin and eosin for histological assessment of fibrosis/cirrhosis grade plus assessment of any ductular reaction, cellular atypia or steatosis, for example. For fibrosis quantification, four sections should be stained with Sirius Red with nine random images taken per slide for a total of 36 images that can be analysed by the BIO-QUANT Life Science morphometry system®. Alternatively, hydroxyproline quantification can be performed. Col1A1 mRNA can be assessed by qRT-PCR and protein by Western blot. Portal pressures can be assessed at the time of sacrifice by inserting a 16 G angiocatheter into the portal vein and measuring the height of a water column. Biometric assessment In a typical experiment an experimental drug is concomitantly administered along with TAA. A possible readout system to assess the efficacy of the drug is the mRNA expression of Col1A1 which may change at the end of the experiment by a factor of 8 (with an inter-individual variance of 25%) (cf. Figure 1d). The control group will receive only TAA. It can be estimated that the drug may reduce the expression of collagen by a factor of 2. In addition, experience in our laboratories has shown that a difference of less than 30% in collagen mRNA expression does not necessarily impact intrahepatic collagen deposition and scarring. For such an experiment, in our view it is advisable to use nine animals per group. If the mortality rate of TAA when applied for eight weeks is further assumed to be approximately 10%, this experiment should be planned with 10 animals in each group. For details on how to calculate biometric parameters, please refer to Scholten et al.’s article ‘The carbon tetrachloride model in mice’ published in this special issue of Laboratory Animals.
Practical implementation
We provide here two TAA protocols, by IP injection in Sprague–Dawley rats and oral administration in C57BL/6 mice. We have found these approaches to produce reliable and reproducible fibrosis and cirrhosis that allows for an easy distinction between control and treatment groups.
TAA via IP injection
Our experience with IP injection of rats with TAA was recently published
14
and is summarized below as a standard approach for reference:
Animals: Sprague–Dawley rats between 280–300 g purchased from The Jackson Laboratory (Bar Harbor, ME, USA) are kept in a 12 h light–dark cycle with free access to water. It should be noted that recent guidelines from the US National Institutes of Health (NIH) suggest equal numbers of male and female animals be used in all future experiments. TAA preparation and administration: TAA purchased from Sigma Chemical Co (St Louis, MO, USA) is dissolved in 0.9% normal saline to a total volume of 1–1.5 mL per rat approximately one hour before injection. Treatment groups receive 150 mg/kg of TAA three times per week for a total of 11 weeks while control groups receive the same volume of 0.9% normal saline. General handling: Rats are examined regularly for signs of distress or ill health and weighed prior to each injection to accurately calculate the dosage of TAA. Injection technique: Rats are handled at all times with care, as the experience can be distressing. We recommend that lab personnel with specific training in animal handling be responsible for the IP injections. Rats are scuffed behind the neck between the middle and index fingers with the tail hooked between the little and ring fingers to expose the abdomen. A sterile, disposable syringe (25–27 G) is used to inject the TAA into the lower side of the abdomen with care taken to avoid injection into the bladder or abdominal organs. The solution should be at room temperature at time of injection. The routine administration of anaesthesia is generally not recommended as it results in unnecessary stress to the animals. Animal sacrifice: At the end of the study period animals are placed under inhalation anaesthesia with 1–5% by volume isoflurane plus analgesia and laparotomy performed. Portal pressures can be measured as described prior to the removal of organs. Side-effects: TAA administration can lead to cirrhosis and its associated complications; however, these are rarely the source of any discomfort to the animals. Mild body weight loss is likely to be observed. Mortality of up to 15% can be expected in the treatment groups. This should be reported in the Methods section of any publications. Efficacy of treatment: After 11 weeks of treatment at the dose of 150 mg/kg three times per week, virtually all of the animals will have developed the histological features of cirrhosis.
TAA via oral administration
Our experience with orally administered TAA was recently published
13
and is summarized below as a standard for reference:
Animals: 8–10-week-old C57BL/6 mice purchased from The Jackson Laboratory are kept in a 12 h light–dark cycle with free access to water. It should be noted that recent guidelines from the US NIH suggest equal numbers of male and female animals be used in all future experiments. Of note, male mice have been reported to be more susceptible to development of TAA-induced liver fibrosis,
33
which is in line with similar findings in other fibrosis models. TAA preparation and administration: TAA purchased from Sigma Chemical Co (St Louis, MO, USA) is dissolved in drinking water at 300 mg/L and administered for 2–4 months. Control animals are given normal water with the same volume. In order to reduce the risk of infection, drinking water should be filtered and acidified (pH around 4.0). The water volume should be measured on a daily basis and changed twice weekly. General handling: Mice are examined daily for signs of distress or ill health. The body weight is determined twice a week starting at the beginning of the experiment. Animal sacrifice: At the end of the study period animals are placed under inhalation anaesthesia with 1–5% by volume isoflurane plus analgesia and laparotomy is performed. Portal pressures can be measured as described prior to removal of organs. Side-effects: TAA administration can lead to cirrhosis and its associated complications; however, these are rarely the source of any discomfort to the animals. Mild body weight loss is likely to be observed. Low mortality can be expected in the treatment groups. This should be reported in the Methods section of any publications. In general, oral administration of TAA leads to less extrahepatic toxicity than IP administration due to the first-pass effect. In contrast to the intraperitoneal treatment, mice are not known to experience pain and a more continuous dosing is achieved. Because of these advantages, we prefer oral administration to IP dosage, although the latter has been used more frequently in the literature. Efficacy of treatment: There is a progressive development of fibrosis and cirrhosis development across the study period. At the end of four months virtually all of the treatment animals will have developed the histological features of cirrhosis.
Classification of severity of procedure
According to Article 15 of the EU Directive 2010/63 (http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:276:0033:0079:en:PDF) the estimated degree of pain, suffering, distress or lasting harm to the animals subjected to TAA application should be classified according to a scoring system that consists of ‘non-recovery’, ‘mild’, ‘moderate’ or ‘severe’. The classification criteria that underpin this assessment have been established by the Expert Working Group on severity classification of scientific procedures performed on animals. Further details can be found at: http://ec.europa.eu/environment/chemicals/lab_animals/pdf/report_ewg.pdf.
The severity of the procedure depends on the TAA dose and duration of the experiment. TAA application to induce fibrosis with no major impairment of liver function is classified as a moderate procedure according to Article 15 of the aforementioned EU Directive 2010/63. The application of TAA to induce liver failure with associated complications such as cerebral oedema and encephalopathy, coagulopathy, renal, haemodynamic and cardiorespiratory failure is classified as a severe procedure. According to the 3 R principle, the procedure should undergo a refinement, and humane endpoints must be implemented with frequent observation points to restrict pain, suffering, distress or lasting harm to the animals, at which point the procedure can be reclassified as moderate.
Concluding remarks
The administration of TAA via the oral or IP route is a safe and effective technique for the study of fibrosis in rodents. There is a wide variation in the route, dose, timing and animal used. The scientific community would benefit from the adoption of a universal SOP for the administration of TAA with the intent of studying fibrosis and its associated complications. Here we have presented two TAA models with which we have had success in reliably and reproducibly induce chronic liver injury and fibrosis in rodents.
Footnotes
Ethical statement
All animal experiments were performed in accordance with the United States Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Institutional Animal Care and Use Committee (IACUC) of the Icahn School of Medicine at Mount Sinai approved all the animal experiments performed.
Funding
Work in Dr. Friedman's laboratory is supported by NIH Grants RO1DK56621 and RO1AA020709.
Conflict of interest statement
The authors have no conflicts of interest to declare.