
Abstract
Metabolic liver injury is one of the fastest growing health problems worldwide. Alcoholic and non-alcoholic fatty livers have been shown to be associated with progression to end-stage liver diseases, as well as to liver cancers, in humans. More importantly, there are no validated therapies for these disorders, therefore intensive research is required in this area. This review of standard operation procedures focuses on the experimental models of fatty liver disease in the mouse. Firstly, use of these experimental models might improve understanding of underlying mechanisms, and secondly this might help to test potential therapeutic options. This article includes, besides a short historic background, an insight into the pathobiochemical mechanisms and detailed experimental procedures as well as the practical implementation of these models.
Historic background of the models
Obesity and obesity-associated conditions including insulin resistance, diabetes mellitus type 2, and hepatic steatosis (fatty liver) are increasing worldwide. Non-alcoholic fatty liver disease (NAFLD), defined as >5% hepatocytes with fatty infiltration, affects approximately 20–30% of the population in Western countries, 1 of whom one-quarter show signs of severe liver dysfunction and/or portal hypertension (varices, encephalopathy, splenomegaly or ascites). 2 Moreover, non-alcoholic steatohepatitis (NASH), defined as NAFLD with concomitant active hepatic inflammation, is therefore the most severe form of NAFLD with a frequency of about 5% to 15%.1,3–5 Within five years NASH patients reach a 40% risk of progressive fibrosis or cirrhosis and around a 5% risk of complications of end-stage liver disease. Significantly, the stage of fibrosis is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up.6–9
In order to mimic this important clinical disorder with its strong economic impact, several dietary models have been described, which induce NASH-like phenotypes in rodents. These nutritional models are either highly caloric in nature or lack essential nutritional components, which lead to shortage-associated symptoms including steatosis and hepatic fat accumulation. Although the applicability of animal models in obesity research is in some cases a controversial and debatable issue10–12 there is no doubt that different experimental models, either genetic and/or dietary, are particularly suitable for investigations aimed at the comprehension of NASH aetiology and development of appropriate regulation. Historically, one of the first models for induction of fatty liver, introduced by Banerji in 1940, was based on a high-fat diet (HFD) that contained large quantities of lard. 13 . Numerous subsequent studies have demonstrated that most HFDs not only induce fatty liver but also promote, to a varying degree, hyperglycaemia and insulin resistance.14–16 Animal models for induction of fatty liver in rats and mice by restriction of choline, methionine, or both, were developed in several experimental studies showing the important function of these components in the maintenance of a normal hepatic function.17–19 The feeding of alcohol as a hepatotoxic high caloric agent in rodents was systematically introduced in 1945, 20 and alcohol-based diets have been subsequently applied worldwide from 1965. 21 Other laboratories apply specialized high fat, high carbohydrate diabetogenic diets with or without cholesterol. Recently, these diets have become very popular because of their capability for altering intestinal microbiota and permeability, leading to chronically increased flooding of lipopolysaccharides into the portal venous blood. These contribute to the induction of inflammation and are possible mediators of insulin resistance, might alter adipose tissue development, and favour hepatic lipid storage in micro and macrovesicular steatosis.22,23
However, all these commonly used nutrients present a high variability of contents, and many suppliers provide chows with different compositions and combinations of ingredients. Additionally, several animal facilities pretreat their nutrients (autoclaving, irradiation, pasteurization) before feeding, potentially destroying individual compounds.24–26 Ignoring the expiration dates of the diets might also be critical since some ingredients have antioxidant properties (such as tocopherols), which are important in protecting oxidation of the most sensible constituents of the diet. This is particularly significant in diets enriched with fatty acids because these molecules are extremely susceptible to oxidative damage. Furthermore, potential unidentified microbial contaminations and any kinds of impurities within the diet might have a critical impact on the outcome of an individual experiment. All these issues related to dietary standards for laboratory animals were officially individuated in 1969 27 . Therefore, special advisory committees were constituted, which were responsible for the discussion of most relevant themes in the field and the charge of assigning specific guidelines. 28 However, a precise definition for the term ‘high-fat diet’ is still missing, and a multitude of different high caloric diets, with fat percentages varying between 20% and 60% of energy as fat originating from lard, beef tallow, fish oil, milk fat, or plant oil, are currently used. 29 Therefore, a consequent heterogeneity of published data emerging from the employment of such a wide range of different variables is not surprising. Moreover, certain mouse strains are more prone to develop obesity (e.g. AKR/J, DBA/2J, C57BL/6J) than others (e.g. C3H/HeJ, BALB/cByJ, C57L/J) and prefer diets with more fat when given a free choice of macronutrients. 30 Lastly, it should be noted that genetic manipulation of the enzymes involved in lipid homeostasis or mobilization may directly influence hepatic fat metabolism or deposition in mice.31–34 Based on these considerations, we attempt to provide some general recommendations in adopting different models used to induce fatty liver in mice.
Pathogenic mechanisms of liver damage
High-fat diets
As mentioned above, the first studies on the effects of HFDs in rodents were performed in the middle of the last century.13,35 Thereafter, several different variations in the composition of HFDs were developed, resulting in a lack of standard parameters for its use in experimental conditions. 29 . Therefore, it is difficult to compare the results of many studies, and this sometimes leads to misinterpretation of the data in regard to hepatic fat accumulation, serum biochemical parameters, histopathological outcomes, development of insulin resistance, obesity and dyslipidaemia, which can be further influenced in rats and mice by genetic background and gender.36–46 This assumption was confirmed in a highly systematic analysis in which metabolic and molecular effects of different HFDs were compared by microchip analysis, which showed that metabolic changes and the resulting phenotypic alterations that are induced by feeding individual fat components are highly variable. 47
Novel variations of the classic HFD are represented by ‘Western diets’, which have a high-fat content and high concentrations of carbohydrates (sucrose or fructose up to 45% weight) which aim to reproduce human high caloric fast food feeding. 48 Moreover, in low density lipoprotein (LDL) receptor-deficient mice it has been demonstrated that high cholesterol supplementations alter hepatic phenotype, suggesting that dietary cholesterol confers a further hit that exacerbates hepatic macrophage infiltration, and increases hepatic inflammation and free oxidative radical generation, thereby representing an important risk factor for hepatic steatosis and progression to steatohepatitis. 23 However, the applied concentrations of cholesterol are highly variable in different studies, which limits the comparison of results or translation to the human situation.
Methionine and choline deficient (MCD) diet
Several compounds such as methionine and choline cannot be synthesized at all, or only in amounts that are necessary to guarantee normal body function, and must therefore be supplied with the diet. The sulphur-containing methionine for example is an essential amino acid that cannot be synthesized de novo and is essential in the formation of S-adenosylmethionine (SAM) that serves as a methyl donor. It is an indispensable compound for the synthesis of cysteine, lecithin, phosphatidylcholine and many other macromolecules (Figure 1). Shortage in methionine is therefore associated in rodents with a progressive physiopathology characterized by increased oxidative stress, significant up-regulation of proinflammatory and profibrotic genes in liver, increase of hepatic aminotransferases and manifestation of other NASH-associated symptoms.
49
Likewise, choline (formerly classified as Vitamin B4) is an integral part of phospholipids (e.g. phosphatidylcholine and sphingomyelin) and a precursor for acetylcholine. Studies in rats have shown that restriction of choline intake results in hepatic lesions that are associated first with severe steatosis and elevated cell turnover, while at later stages clearance of deposited fat, fibrosis and parenchymal nodularity occurs.
50
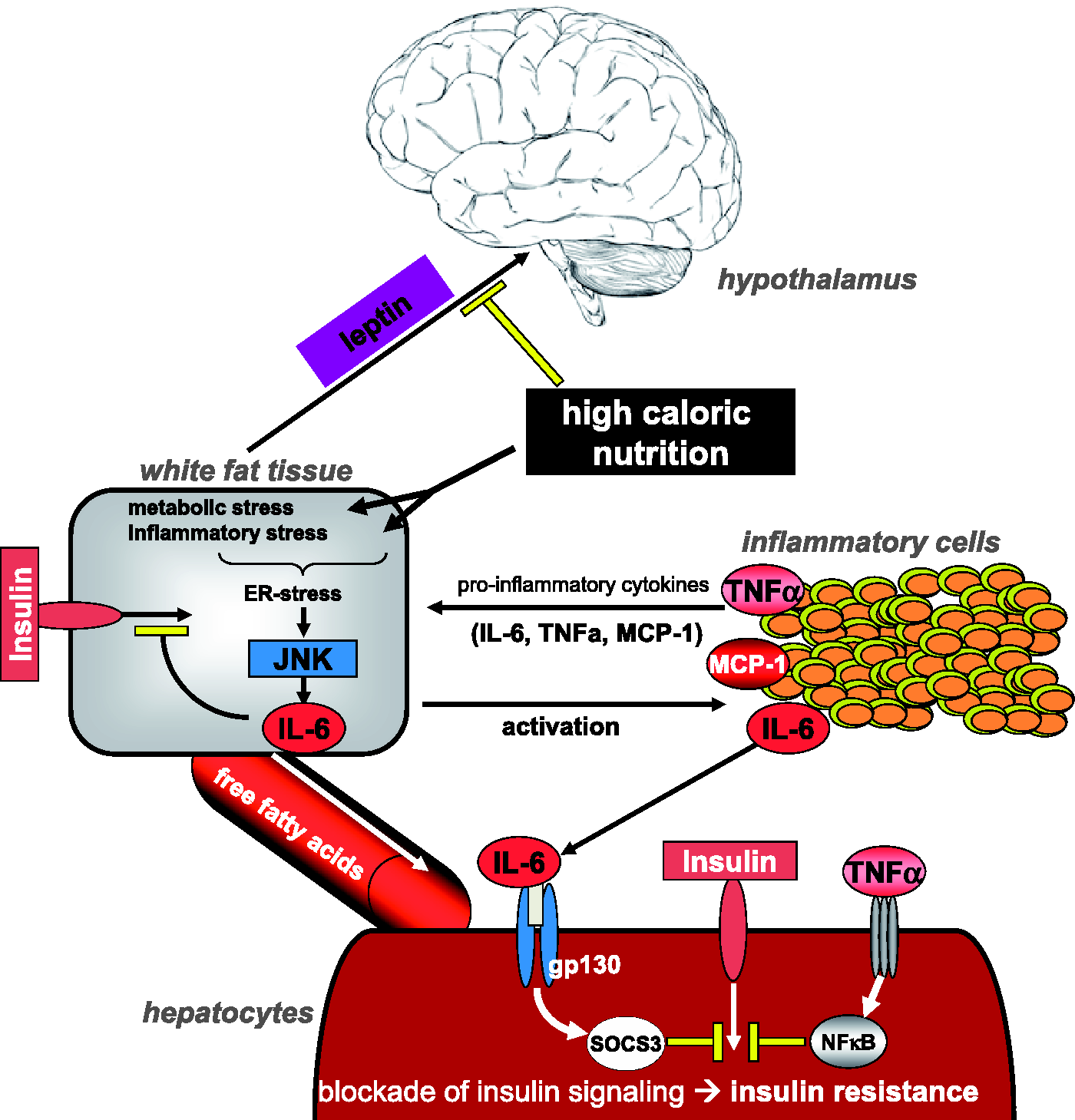
The MCD diet, that lacks these two essential nutrients, causes rapid micro and macrovesicular steatosis. In particular, it promotes intrahepatic lipid accumulation through a decreased production of very low density lipoproteins (VLDL), a drastic down-regulation of key enzymes involved in triglyceride synthesis (as SCD-1), and an impaired de novo lipogenesis. As a consequence, this diet leads to increased reactive oxygen species (ROS) generation and inefficient β-oxidation resulting in hepatocytes ballooning and diffuse necrosis, finally evolving to hepatic fibrosis.51–53 Mechanistically, it is most likely that reduced VLDL-mediated export of triglycerides and increased lipolysis of the adipose tissue are the major contributors to the development of hepatic steatosis.54,55 During the inflammatory response, activated macrophages infiltrate the liver and upregulate critical proinflammatory pathways and mediators such as NF-κB, ICAM-1, COX-2, MCP-1, TNF-α, and IL-6 (Figure 2).56–58 Compared with human NASH, however, it is well known that the MCD model in rodents results in lower plasma triglyceride and cholesterol levels, weight loss, and a metabolic profile that is the opposite to that observed in humans54,23 (Figures 2 and 3).
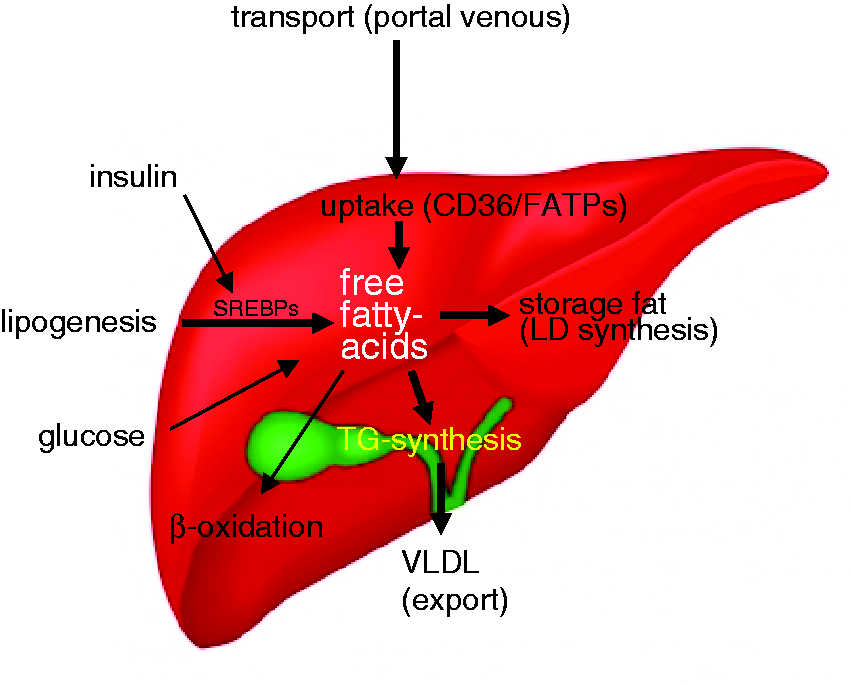
Essential dietary supplements. Methionine is an essential sulphur-containing proteinogenic amino acid that is also part of the methyl group donor S-adenosylmethionine (SAM), which is indispensable in the synthesis of various nucleic acids, proteins, lipids, and secondary metabolites. Choline is an integral part of phosphatidylcholine and sphingomyelin, and a precursor for the synthesis of the neurotransmitter acetylcholine. Non-alcoholic steatohepatitis (NASH) development. NASH development depends on a complex interplay between regulating structures such as the hypothalamus, the physiology of white fat tissue and the recruitment of immune cells to the liver. Certain adipokines and cytokines thereby fulfil different important regulatory functions. Free fatty acid metabolism. Intrahepatic metabolism of free fatty acids in the liver are controlled by numerous mechanisms, such as dietary intake, intrahepatic storage, β-oxidation, and also the subsequent export of newly synthesized fatty acids.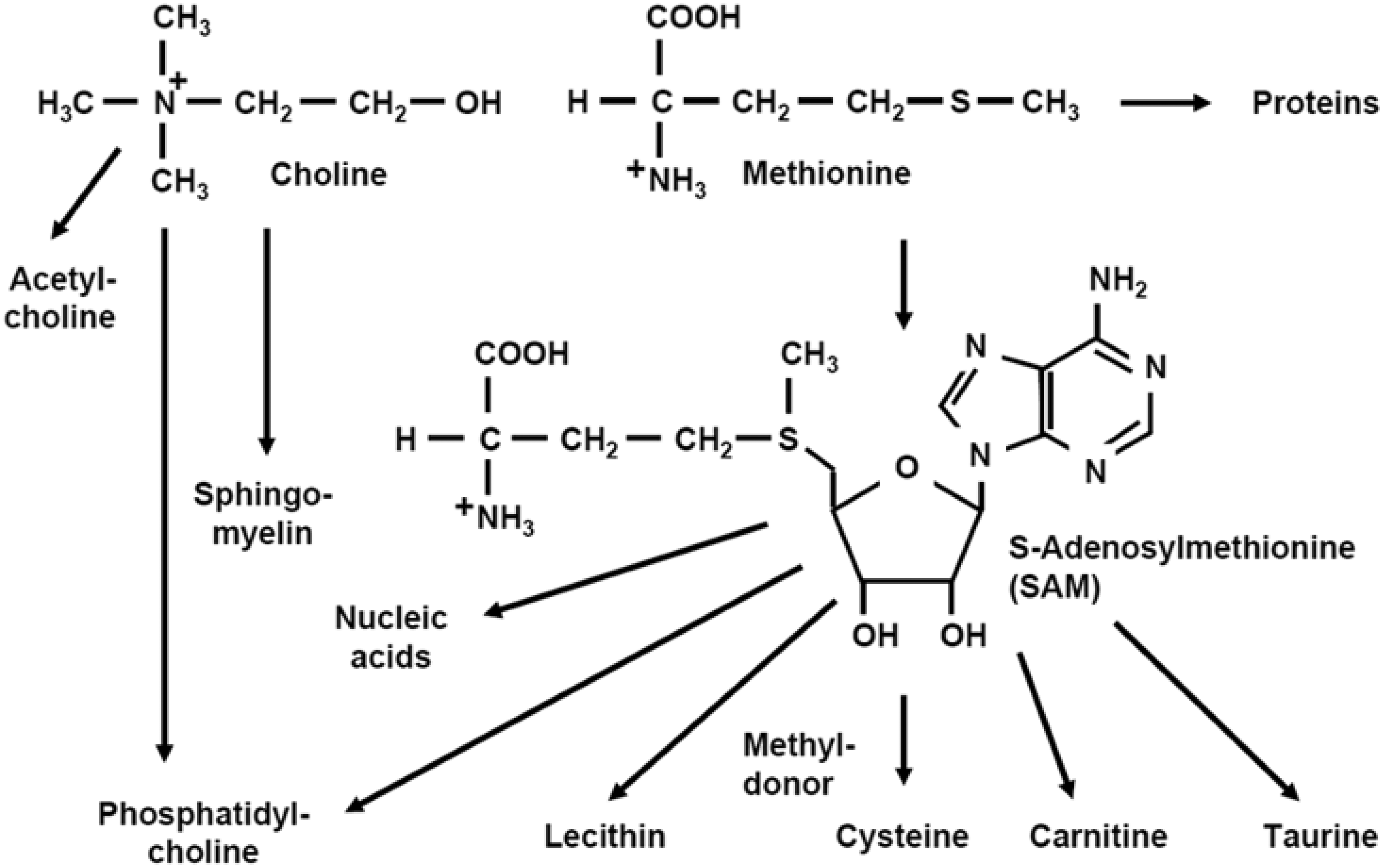
Choline deficient (CD) diet
As mentioned above, insufficient supply of choline results in hepatic alterations. The curative effects of choline in CD rats and mice have been shown decades ago.17,18 In addition, rats fed a CD diet for short periods (2 weeks) show threefold higher amounts of trigylceride and esterified cholesterol in hepatic lipids, while the composition of individual fatty acids varies in a sex-specific manner. 59 The concentration of choline that is necessary to maintain a physiological hepatic lipid content and a normal lipotropic activity is dependent on temperature and is inversely correlated to the content of methionine and proteins.60,61 Compared to the MCD diet, CD feeding results in a less severe phenotype that is characterized by microvesicular steatosis, mild hepatic inflammation, obesity, dyslipidaemia, and insulin resistance. 62 All these changes are associated with exacerbated mitochondrial oxidative injury affecting mitochondrial lipids, proteins, and concentration of the adenosine triphosphate (ATP) synthase resulting in lower quantities of hepatic ATP. 63
3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet
A well-established model for the induction of sclerosing cholangitis and a biliary type of liver fibrosis in mice is induced by feeding a DDC diet. It has been used in the past to study the formation of Mallory bodies, which are hepatocellular inclusion bodies characteristically associated with alcoholic and non-alcoholic steatohepatitis, metabolic liver diseases (such as Wilson's disease and other forms of copper toxicosis), and chronic cholestatic liver diseases. 64 The model involves the secretion of proinflammatory mediators and cytokines such as TNF-α and IL-6 and the activation of profibrogenic factors such as platelet-derived growth factor and transforming growth factor-β. Together with increased biliary porphyrin secretion, this leads to activation of biliary epithelial cells, giving rise to a significant pericholangitis. As a consequence, the secretion of toxic metabolites is impaired, further triggering hepatocyte injury and inhibition of tissue repair. In addition, DDC feeding activates the intrahepatic stem cell compartment. Oval cells are induced and start to proliferate mainly because hepatocytes are unable to enter the cell cycle. In consequence, proinflammatory cytokines such as TNF-α 65 and the related factor LT1b 66 are essential to trigger oval cell activation during toxic liver injury.
Alcohol and Lieber-DeCarli diet
Rats and mice are the most appropriate species when models of alcohol abuse are being investigated. The first well controlled demonstrations that selective and highly controlled feeding of ethanol to rodents results in a progressive rise of total lipids, hepatic accumulation of triglycerides, increase in liver weight and histomorphological changes were first reported in the years 1949–1965.20,21 In the original protocols, alcohol was either given as a 15% aqueous solution as a replacement of drinking water 20 or alternatively administrated in five or six daily doses by oral gavage. 21 Subsequently, different standardized variations of the Lieber-DeCarli liquid diet using alcohol in combination with nutritionally adequate diets were proposed. These combined diets represent an experimental model much closer to human conditions and more suitable for mimicking different clinical settings of fatty liver and related hepatic alterations.67,68 In other protocols, alcohol is preferentially administered ‘ad libitum’ in drinking water for variable time intervals and in variable concentrations. A recent published protocol described chronic alcohol administration with a single binge ethanol feeding synergistically which induces liver injury, inflammation and fatty liver, and which mimics acute-on-chronic alcoholic liver injury in patients. 69 Most of these protocols have turned out to induce reproducible features of hepatic steatosis but without further progression to bridging hepatic fibrosis and end-stage cirrhosis.70,71 However, when ethanol is applied in combination with cancerogenic agents such as diethylnitrosamine, it significantly increases tumour incidence in male but not female mice. 68 However, all these alcohol models have some limitations and caution should be applied in the interpretation of the data. 70 Indeed, by contrast with the human situation, the consumption of alcohol does not permanently increase over time and the rate of alcohol catabolism is up to five times higher in rodents, 72 even though it induces remarkable oxidative stress in zones 1–3 hepatocytes as reviewed. 73
General remarks
The intrahepatic free fatty acid metabolism in humans and animal models is highly complex. The concentration of free fatty acids in the liver is mainly affected by controlled dietary intake, intrahepatic storage, β-oxidation and also the subsequent export of newly synthesized fatty acids (Figure 3). Notably, the most important risk factors for NAFLD in humans are male gender, age, obesity, insulin resistance and cardio-metabolic alterations that define the metabolic syndrome. 1
Moreover, several genetic mouse models have been shown to provide a base for the development of fatty liver diseases and NASH development (as discussed in the following section).
Experimental procedure
General considerations
Genetic background
It is well accepted that the susceptibility towards metabolic induced liver injury in mice is strongly strain-dependent. In particular, AKR/J, DBA/2J, C57BL/6J inbred mice are more prone to developing metabolic induced hepatic changes than others (e.g. C3H/HeJ, BALB/cByJ, C57L/J). An interesting model for the study of metabolic changes is a mouse carrying a deficiency in the hormone leptin (ob/ob) and its receptor (db/db). Leptin usually lowers energy uptake and consumption, thus physiologically preventing the development of excess weight. Mice of the ob/ob strain are especially prone to develop obesity, NASH and insulin resistance. 74
Other important players in the lipid metabolism, apart from leptin, are apolipoproteins (Apo), especially ApoE, whose genetic deficiency has been shown to lead to atherosclerosis, hypercholesterinemia and obesity. 75 The administration of a Western diet (a high-fat, cholesterol-rich diet) in these mice promotes the development of NASH with fibrosis within the features of the metabolic syndrome in only seven weeks. 76
Interestingly, alterations of the renin–angiotensin system in rodents have also been shown to induce NAFLD in mREN-2 transgenic rats, harbouring mouse renin-2 gene in their genome. 77 These rats spontaneously develop hepatic steatosis and inflammation at the age of 12 weeks.
Morphological analysis and quantification
Non-alcoholic fatty liver disease activity score (non-alcoholic steatohepatitis).

Activity scores are defined as: 0–2 definitive no steatohepatitis; 3–4 possible steatohepatitis; > 5 definitive steatohepatitis.
Specific practical implementation
General points
All the described methods adopted for challenging the metabolic status of a rodent organism are based on either changed proportions of individual nutritional components or the addition of distinct chemicals to the chow. Thus, all the metabolic animal models above indicated are based on feeding different chows and do not involve further chemical administration or other physical handling of experimental animals.
Animal handling and husbandry
Before treatment mice are weighed and examined for signs of distress (i.e. changes in respiration, rough hair coat, unusual behaviour, or hunched posture). Animals should be periodically inspected for abnormalities during the time of treatment and those showing behavioural alterations or signs of sickness should be excluded from investigations. At the end of the experimentation (e.g. after 6–24 weeks) animals are sacrificed by cervical dislocation.
Specific points related to individual models
Content of dietary chows (% weight).
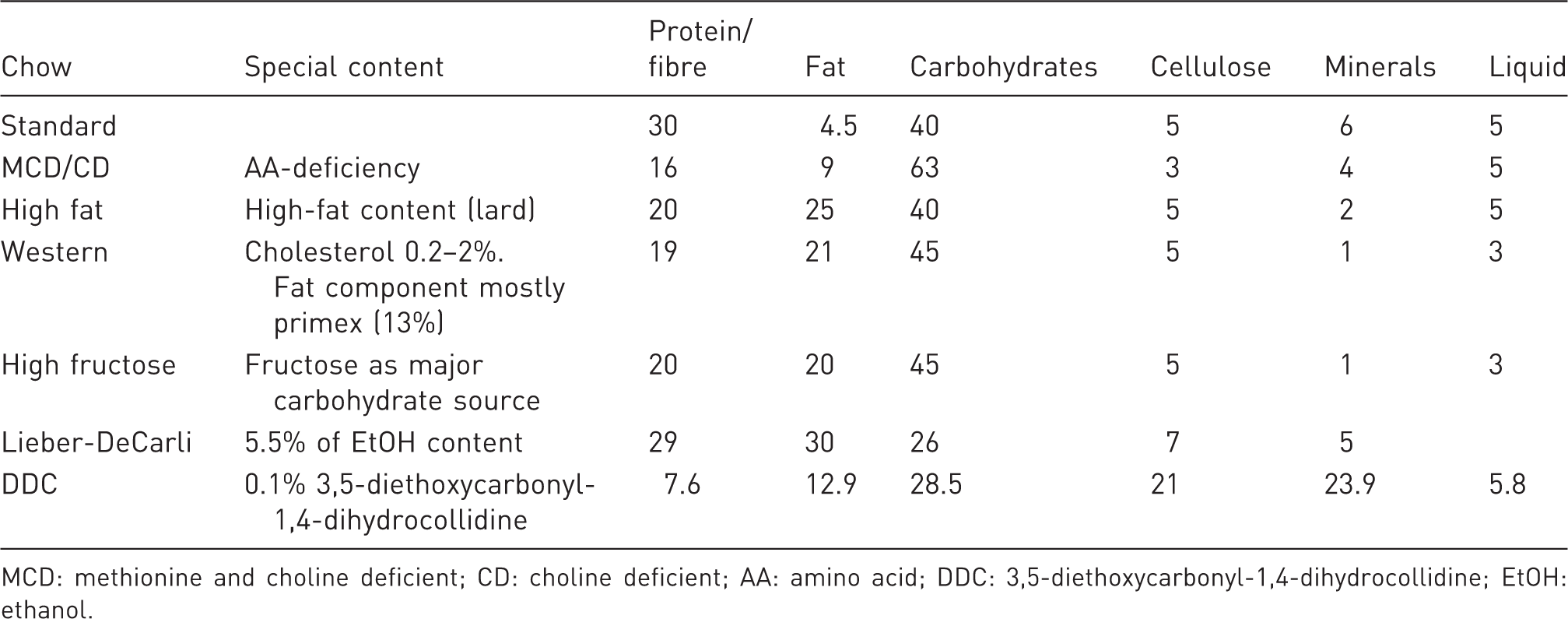
MCD: methionine and choline deficient; CD: choline deficient; AA: amino acid; DDC: 3,5-diethoxycarbonyl-1,4-dihydrocollidine; EtOH: ethanol.
High-fat diet
Mice usually accumulate abdominal fat and liver steatosis with a moderate increase in the liver/body weight ratio. No obvious complications are registered other than a light hyperglycaemia which is not always correlated with insulin resistance. Indeed, heterogenic phenotypes due to a greater variability of the individual response to the diet might result in a statistical inconvenience. Furthermore, under classic HFD simple steatosis rarely evolves into steatohepatitis in wild-type mice (Figure 4).
High-fat diet (HFD) and Western diet histology. On the left, mouse liver sections after the feeding of a HFD for four weeks. Histology determined by haematoxylin and eosin (H&E) staining shows moderate lipid droplets accumulation confirmed by Oil-Red-O (ORO) staining (red). On the right, mouse liver sections after the feeding of a Western diet for 20 weeks. Top: H&E stain illustrate massive macrosteatosis (long arrows) mainly surrounding the portal spaces (PS). Short arrows indicate microsteatosis predominantly located around the perivenular areas (PV). Bottom: ORO staining confirms abundant lipid droplets accumulation (red).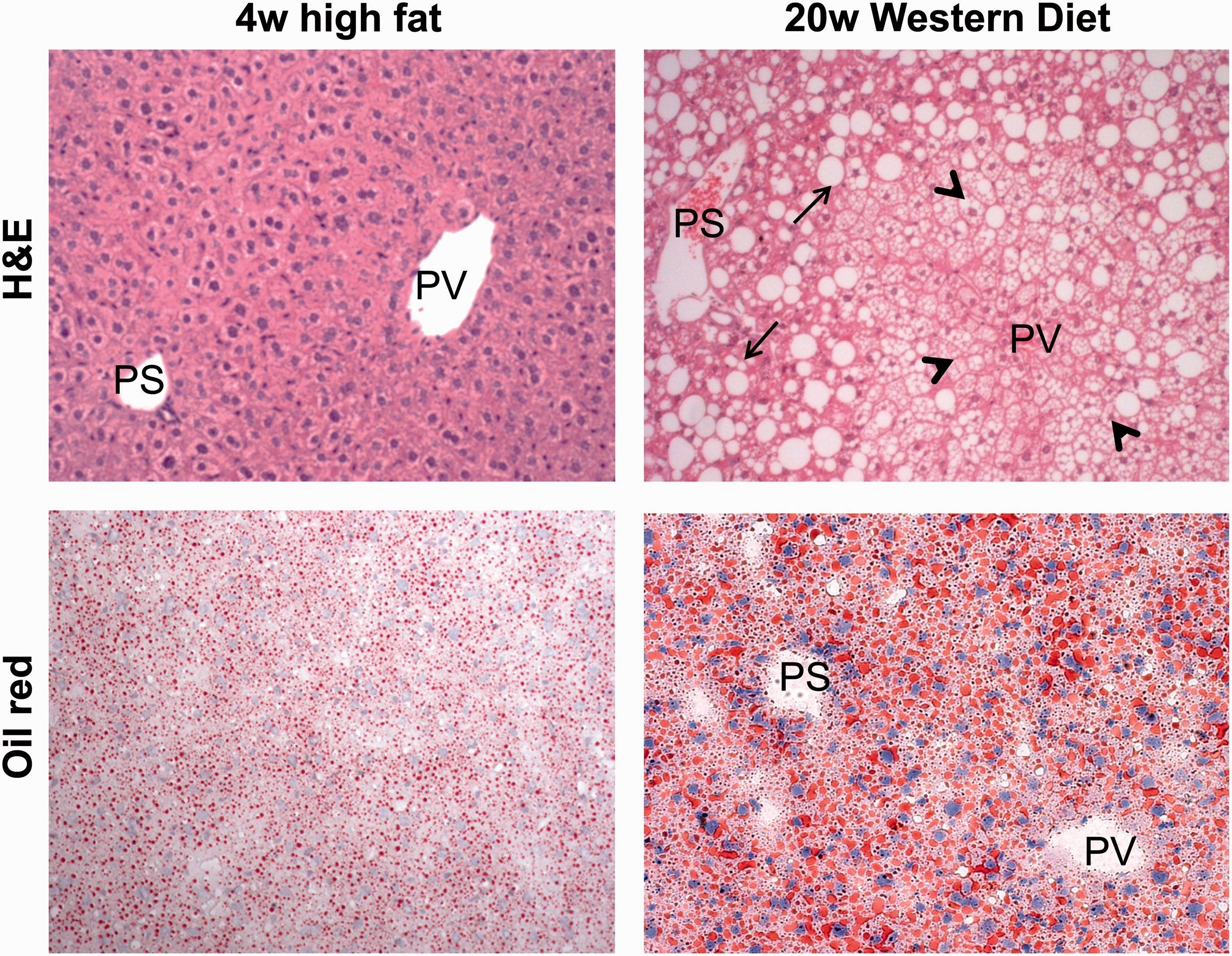
Treatment duration: 12–24 weeks.
High fructose diet/Western diet
Recent studies indicate that metabolites deriving from the assumption of high fructose concentrations (corn syrup derivatives) in combination with high fat can further exacerbate fat accumulation and inflammatory response. High fructose feeding of long duration (up to 30 weeks) typically results in steatohepatitis progressing to moderate fibrosis. Increasing cholesterol concentrations in the diet (from 0.2% to 2%) has been shown to accelerate the degree of fibrosis due to a stronger impact on macrophages and stellate cell activation. 79 After prolonged feeding periods mice are expected to gain weight and develop obesity relatively quickly, followed by stabilization of body weight. Interestingly, mice develop a more dramatic hepatomegaly than with administration of a classic HFD.
Treatment duration: 16–30 weeks.
MCD diet
Mice subjected to MCD treatment suffer from the complications of liver injury and a general wasting syndrome. A progressive decrease of the liver/body weight ratio is however associated with an early appearance of macrosteatosis and massive inflammatory infiltrate (Figure 5). They show loss in body weight and progressively decreased food intake, and are more fatigued, however they do not experience pains or mutilations. Mice should be weighed and withdrawn from an experiment, if their weight drops by more than 20% of their starting body weight and remains so for more than 48 h.
Methionine and choline deficient (MCD) histology. Haematoxylin and eosin and Oil-Red-O staining of mouse liver sections after four weeks of MCD treatment. Massive macrosteatosis accompanied by clusters of inflammatory cells, mainly CD11b+, is observed.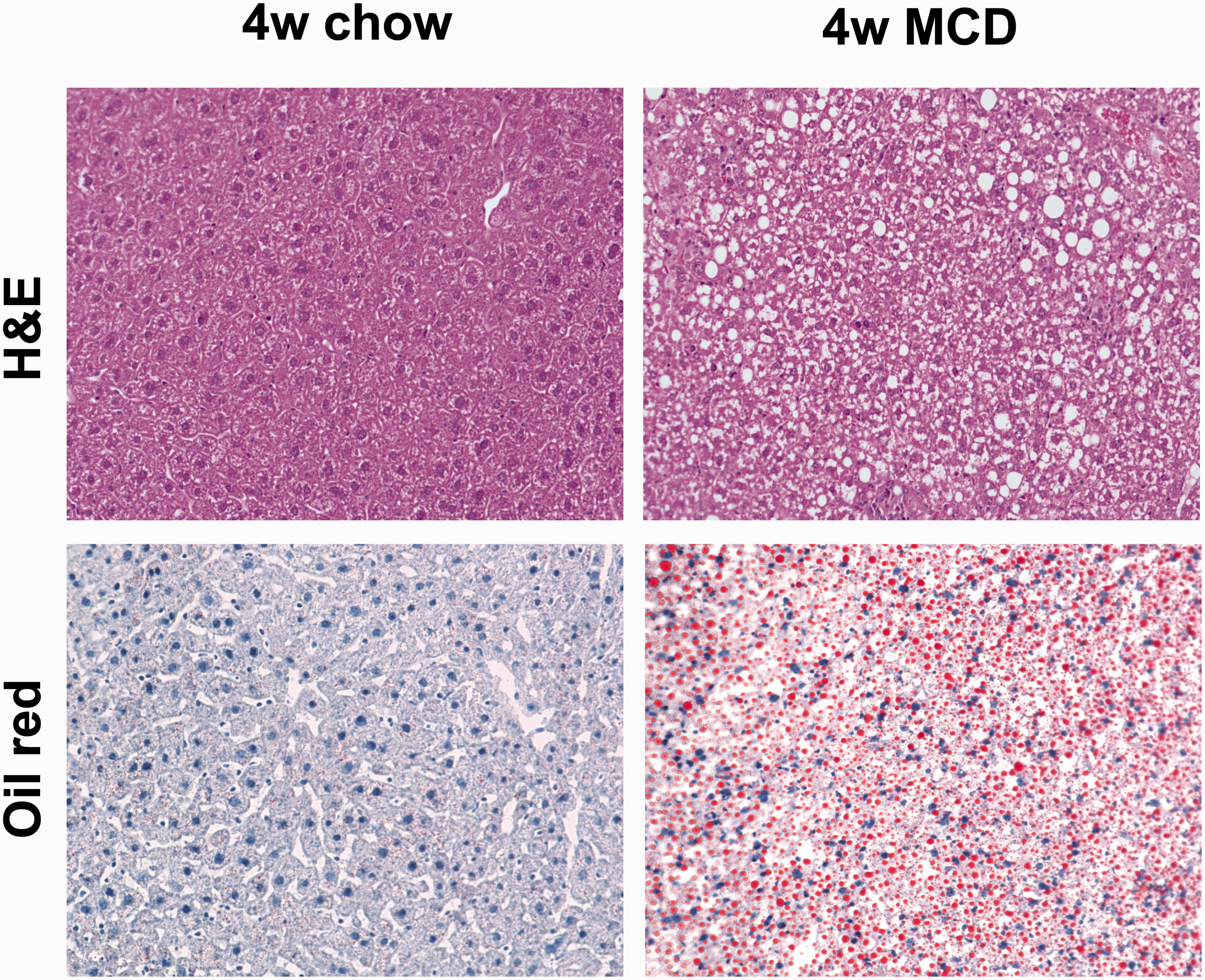
Treatment duration: 4–10 weeks.
Ethanol (Lieber-DeCarli) diet
The Lieber-DeCarli diet is a well-established rodent model of alcoholic steatohepatitis. Mice deprived of food can easily consume the diet through drinking water and/or isocaloric control diets. The lack of solid food might represent a problem for rodents because of tooth growth. Within four weeks of feeding, mice develop moderate fat accumulation usually accompanied by inflammatory infiltrate and macrophage activation (Figure 6).
Histology after Lieber-DeCarli diet. Mouse liver sections after feeding with a Lieber-DeCarli diet for four weeks. Histology (haematoxylin and eosin) shows pronounced microvesicular steatosis and moderate inflammatory infiltrate.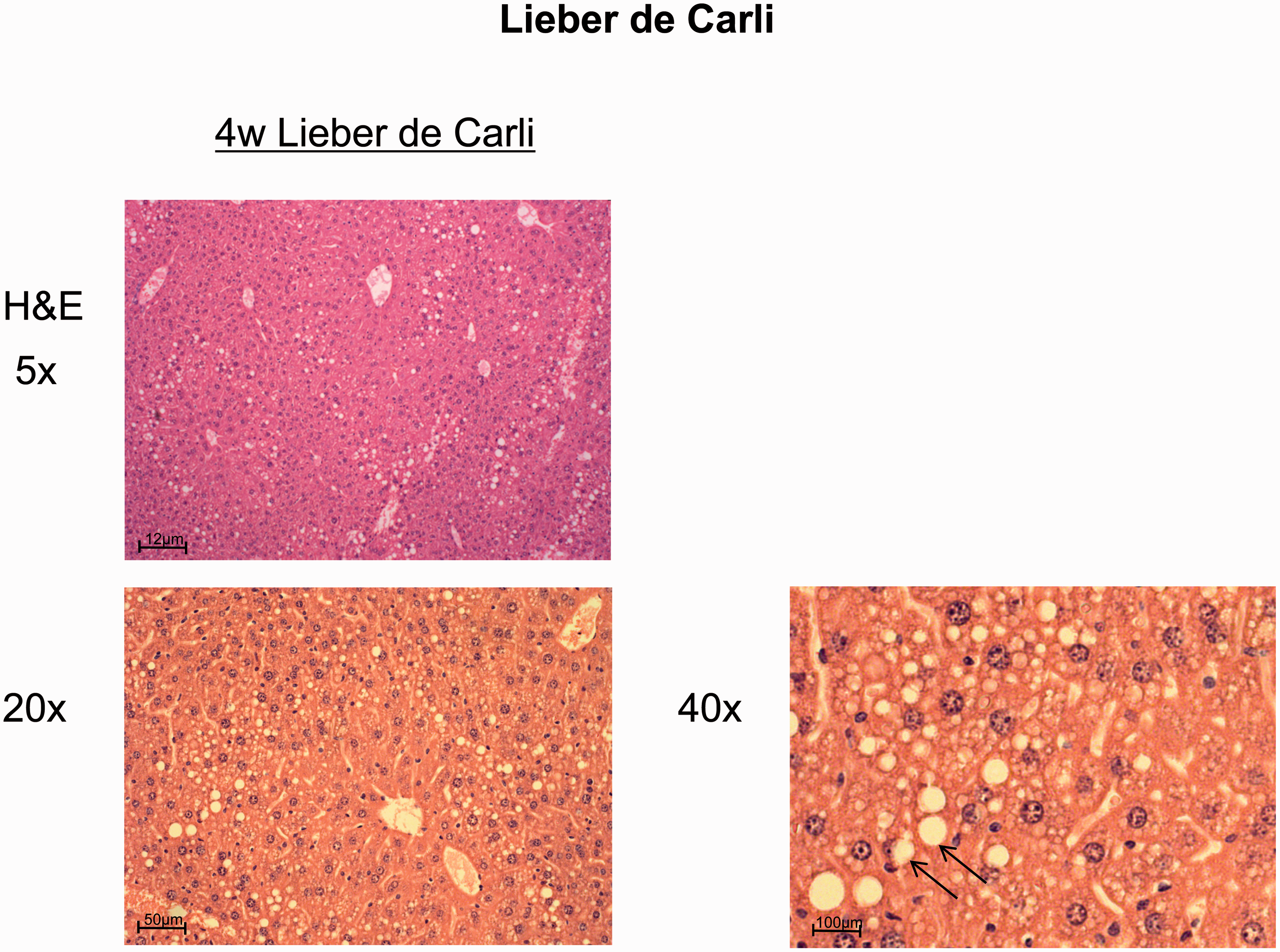
Treatment duration: 4–8 weeks.
DDC diet
DDC treatment induces a rapid onset of liver injury, cholestasis and fibrosis. Therefore, especially at early time points, animals can potentially show loss in body weight and more fatigue. Death of experimental animals can occasionally occur but is rare. Therefore mice are continuously monitored (every 24 h) for their physical health. Mice should be weighed and withdrawn from an experiment, if their weight drops by more than 20% of their starting body weight.
Treatment duration: 4–24 weeks.
Classification of severity of procedure
According to Article 15 of the EU Directive 2010/63 (http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:276:0033:0079:en:PDF) the estimated degree of pain, suffering, distress or lasting harm of the animals subjected to DENA or DMN application should be classified following a scoring system of ‘non-recovery’, ‘mild’, ‘moderate’ or ‘severe’.
Details about the classification criteria that underlie this assessment have been established by the Expert Working Group on severity classification of scientific procedures performed on animals. These can be found at: http://ec.europa.eu/environment/chemicals/lab_animals/pdf/report_ewg.pdf.
The HFD and the high fructose/Western diets as well as the diets containing ethanol (Lieber-DeCarli diet) with no major impairment of liver function are classified as moderate procedures according to Article 15 of the above-mentioned EU Directive 2010/63.
The DDC and MCD diets with no major impairment of liver function are classified as moderate to severe procedures according to Article 15 of the above-mentioned EU Directive 2010/63. In line with the 3R principle these procedures should undergo a refinement, and humane endpoints must be implemented with frequent observation points to restrict pain, suffering, distress or lasting harm to the animals. Then, the procedure can be reclassified as moderate.
Concluding remarks
To date numerous animal models of NASH exist. However every model has its specific aspects and limitations, therefore none of the existing models developed so far can fully capture all the features of human fatty liver diseases. The widely used MCD model results in severe steatohepatitis, but mice – by contrast with humans – lose weight and lack insulin resistance. Other models such as HFD-based feeding strategies mimic changes associated with metabolic syndrome better, but have less inflammatory activity. Thus, one has to choose the appropriate model for both specific application and research interest, as is described and discussed above.
Ethical statement
The paper is a review of the literature. The experimental data shown have been approved by LANUV (Recklinghausen, Germany).
Footnotes
Acknowledgements
We are particularly thankful to Dr Yulia A Nevzorova and Dr Fabienne Schumacher for their kind concession of histological pictures regarding Lieber-DeCarli and HFD mouse feeding. All authors contributed equally to this work.
Declaration of conflicting interests
The authors declare no conflict of interest.
Funding
Deutsche Forschungsgemeinschaft funded the study within the Transregional Collaborative Research Center ‘Organ Fibrosis: From Mechanisms of Injury to Modulation of Disease’ (SFB/TRR57).