
Abstract
Ornithine decarboxylase (ODC), a regulatory enzyme of polyamine biosynthesis, is involved in cell growth and differentiation. Lack of information about the exact cellular and subcellular localization of ODC is one of the main obstacles to precise interpretation of the biological roles of the ODC/polyamine system. Here we describe the development and optimization of an immunocytochemical method to detect ODC in cells and tissues. For this purpose a monoclonal antibody (MP16-2) against a defined epitope of ODC protein was developed. Specificity of the antibody for ODC was substantiated by Western blotting and ELISA analysis using cell and tissue homogenates. In cultured cells, optimal staining results were obtained after fixation with crosslinking fixatives followed by permeabilization with methanol. In rat tissues, ODC immunoreactivity was best preserved in paraffin sections fixed with Bouin's fixative. Antigen retrieval using SDS and citrate buffer substantially increased ODC immunostaining and decreased background staining. Localization studies of ODC in different cell lines showed that strongest staining for ODC was found in the nucleoplasm of mitotic cells, whereas confluent cells showed moderate perinuclear staining. Immunocytochemical studies of various rat tissues showed high cytoplasmic immunostaining of ODC in epithelial cells of kidney, prostate, and adrenal medulla of testosterone-treated rats, in glandular epithelium of small intestine, and in pancreas of neonatal and adult rats.
ORNITHINE DECARBOXYLASE (ODC, EC 4.1.1.17) is the initial and regulatory enzyme of polyamine synthesis, converting the amino acid ornithine into the polyamine precursor putrescine. Polyamines are organic bases that play an essential role in cell growth and differentiation (Pegg 1986). The enzymatic characteristics of ODC have gained considerable interest in various biomedical fields and have been summarized in several reviews (Russell 1985; Persson et al. 1988; Hayashi et al. 1996). The enzyme exhibits rapid and diverse changes in biological activity as an early response to the effect of virtually all growth-promoting stimuli, including hormones, drugs, growth factors, mitogens, and tumor promoters on cells. Recent studies have shown that overexpression of ODC can cause cellular transformation, suggesting that the enzyme can be considered as a proto-oncogen product (Auvinen et al. 1992,1997; Tamori et al. 1995; O'Brien et al. 1997; Shantz and Pegg 1998; Tabib and Bachrach 1998).
Despite extensive biochemical and molecular biological research, the precise role of the ODC/polyamine system in cell physiology remains to be clarified. Insight into the exact cellular and subcellular localization of ODC might provide valuable information on the role of this metabolically important enzyme. Efforts to assess the (sub)cellular localization of ODC by immunocytochemical means have been made in the past (our unpublished work). However, different localization patterns were found, depending on the cell system studied and the antibody used. These patterns varied from a cytoplasmic or (peri)nuclear pattern to a both cytoplasmic and nuclear localization. An explanation for these discrepancies may be the lack of specificity of the ODC antibodies that were used. Further more, effects of cell and tissue preparation (e.g., fixation procedures, permeabilization, dehydration, and embedding in paraffin) on the preservation and/or accessibility of the ODC protein for the antibodies were hardly defined.
To develop a reliable method for immunocytochemical detection of ODC, we raised a specific and potent mouse monoclonal antibody (MP16-2) to a synthetic hexadecapeptide (P16) corresponding to a computer-calculated epitope of ODC (amino acid residues 345-360) (Schipper et al. 1993). The purpose of the present study was to investigate systematically the applicability of the MP16-2 antibody for immunocytochemical detection of ODC in cells and tissues. For this purpose, we further characterized the specificity of the antibody for ODC by Western blotting and ELISA analysis using cell and tissue homogenates. To determine how the ODC epitope was preserved during the consecutive steps of the immunocytochemical procedure, model studies were performed using artificial preparations of P16-coupled Sepharose beads as well as intact cells (human amnion WISH cells). Subsequently, localization studies were performed on cultured cells (WISH cells, HCT 116 human colon cancer cells, ODC-overexpressing NIH 3T3 mouse fibroblasts) and on rat tissues, using optimized preparation procedures as established in the model studies.
Materials and Methods
Antibody
A mouse monoclonal antibody (MAb) against a defined epitope of ODC was produced and characterized as described previously (Schipper et al. 1993). In short, a computer-calculated hexadecapeptide (P16) representing the amino acid sequence 345–360 of ODC was coupled to bovine serum albumin and used for production of MAbs. Antibodies were screened in an ELISA using thyroglobulin (Tg) as a protein carrier for P16. One hybridoma cell line, MP16-2, was positive for the P16-Tg conjugate, whereas it did not react with the protein carrier, indicating that MP16-2 was specific for the P16 peptide. The MP16-2 antibody was identified as an Ig1 immunoglobulin recognizing the C-terminal part (corresponding to amino acids IWGPTC of the ODC sequence) of the P16 peptide as determined by epitope mapping. Purified antibody fractions were prepared from hybridoma cell cultures by affinity chromatography (HiTrap Protein G; Pharmacia, Uppsala, Sweden) and stored in stock solutions of 1 mg/ml. P16 peptide was used as an antigen in the artificial model preparations. P16-Tg was also used as an antigen-protein carrier conjugate in the ELISA assays and for preabsorption of MAb MP16-2 for control immunocytochemical studies.
Cell and Tissue Preparations
Human amnion WISH cells (American Type Culture Collection; Rockville, MD), HCT 116 human colon cancer cells (American Type Culture Collection), and NIH 3T3 mouse fibroblast cells (American Type Culture Collection) were cultivated in Dulbecco's modified Eagle's medium (DMEM; Flow Laboratories, Zwanenburg, The Netherlands) supplemented with 10% fetal calf serum (FCS; Integro BV, Zaandam, The Netherlands), 100 IU/ml penicillin, and 100 μg/ml streptomycin (ICN Biomedicals; Costa Mesa, CA). A solution of 0.5 mg/ml trypsin (Difco Laboratories; Detroit, MI) and 0.2 mg/ml ethylene di-amino tetra-acetic acid (EDTA) in PBS was used to detach cells for passage.
To investigate ODC immunoreactivity in ODC-overexpressing cells, NIH 3T3 cells were transiently transfected with an expression construct (12.3 kb) carrying the full-length gene encoding human ODC driven by gut-specific promoter sequences. Transfected cells showed elevated levels of ODC mRNA (two- to 20-fold) and enzyme activity (3.5-to 40-fold) compared with nontransfected cells (manuscript in preparation).
Cell homogenates were prepared for immunochemical analysis (Western blotting, ELISA) by homogenization of 1 × 105 cells in lysis buffer (50 mM Tris, 1 mM EDTA, 0.3% SDS, pH 7.2). After clarification of the lysates by centrifugation at 12,000 X g at 4C for 10 min, the supernatant was used for further analyses.
We purified ODC from kidneys of testosterone-treated NMRI mice as described by Kopitz et al. (1990) to examine whether the anti-P16 antibody recognized native ODC protein. Two hundred kidneys yielded 180 μg of enzyme protein with a specific activity of 28.3 μU/mg of protein (U = mol/min). A relative purification of 4760-fold and a recovery of 41% of cytosolic ODC activity were obtained. A fraction of the purified ODC was labeled with the selective inhibitor 2-(dinuoromemyl)-3,4-di-3H-DL-ornithine ([3H]-DFMO) as described earlier (Kopitz et al. 1990). This preparation contained no detectable ODC activity after labeling with DFMO. The crude fraction of cytosolic ODC, highly purified ODC, and purified ODC fraction labeled with [3H]-DFMO was subsequently applied as antigen for immunochemical studies using Western blotting and/or ELISA.
Western Blotting
Homogenates were boiled with sample buffer [62 mM Tris-HCl, pH 6.8, 10% glycerol, 2% sodium dodecyl sulfate (SDS), 650 mM β-mercaptoethanol, 0.025% bromophenol blue] for 5 min at 100C. The samples and molecular weight markers (Pharmacia) were loaded on 10% SDS-polyacrylamide gels (acryl/bisacryl 29.2/0.8) and subsequently electrotransferred onto nitrocellulose sheets (Millipore; Bedford, MA) in blotting buffer (25 mM Tris-HCl, pH 8.6, 192 mM glycine, 20% methanol, 0.02% SDS).
Blots were washed with PBS containing 0.05% Tween-20 (Merck; Darmstadt, Germany) (PBST) and incubated for 3 hr at room temperature (RT) in PBST containing 5% low-fat milk powder to block nonspecific binding sites. The blots were rinsed in PBST and incubated overnight at RT with MP16-2 antibody (1 μg/ml) in PBST. After washing three times for 10 min in PBST, blots were incubated with secondary antibody, i.e., peroxidase-coupled goat anti-mouse IgG (Dako; Glostrup, Denmark) diluted 1:2000 in PBST. Finally, bound antibodies were visualized using a chemiluminescent detection system (Boehringer Mannheim; Almere, The Netherlands) followed by exposure to radiographic film (Kodak X-OMAT-R) for 10–30 sec.
ELISA
ELISA analysis was performed as described previously (Schipper et al. 1993), with some modifications. Briefly, polystyrene 96-well microtiter plates (Costar; Cambridge, MA) were precoated with 1% poly-L-lysine in PBS for 3 hr at 37C. Plates were then coated with the antigen solution, e.g., dilutions of P16-coupled Tg (5 μg/ml), HCT 116 cell supernatant (2.5 mg/ml), or purified ODC (0.5 μg/ml), and incubated overnight at 4C. Antigen solution was removed and the plates were incubated with PBS containing 1% gelatin for 3 hr at 37C to prevent aspecific binding of the antibody. After washing with PBST, the MP16-2 antibody (1 x μg/well) diluted in PBS containing 2% bovine serum albumin (BSA) (Sigma; St Louis, MO) and 0.1% Triton X-100 (Sigma) was added to each well and incubated for 1 hr at 37C. After rinsing with PBST, incubation with peroxidase-labeled conjugate (RαM-PO; Dako) diluted 1:1000 in PBST was performed for 1 hr at 37C. After rinsing, wells were filled with a solution of the chromogen 5-aminosalicylic acid (1 mg/ml) (Sigma) and H2O2 (0.25%) in 50 mM phosphate buffer (pH 6.0) and incubated for 30 min at RT. Production of the final reaction product was measured by reading the absorbance at 450 nm in an ELISA plate reader (Titertek; Flow Laboratories).
Artificial Model Preparations
To investigate the effects of the immunocytochemical procedure on P16 antigenicity, the so-called defined antigen substrate spheres (DASS) system was used (Capel 1975; Streefkerk et al. 1975). For this purpose, P16 peptide was coupled to EAH Sepharose 4B beads (Pharmacia).
First, the free amino groups of the Sepharose beads were activated with a stoichiometric amount of 6-(1-maleinimido)hexanoic acid 1-succinimidyl ester (MHS; Fluka, Buchs, Switzerland) in 1 ml N,N-dimethylformamide (DMF; Merck) for 30 min. Beads were then rinsed with DMF, treated with acetic acid N-hydroxysuccinimidic ester (AcONSu; Merck) in DMF for 10 min to block nonactivated amino groups and again rinsed with DMF. For the coupling of 1 ml activated beads 5 mg decapped P16 was used. P16 was dissolved in 100 μl DMF/methanol, 14:5 (v/v) and added to the activated beads. After 1 hr, beads were rinsed with PBS and stored at 4C. Intact bead preparations were used for immunocytochemical model experiments. Alternatively, beads were embedded in paraffin and 4-μm-thick sections from the paraffin blocks were mounted on object slides.
Fixation Methods
Crosslinking and coagulant fixatives were tested with the use of artificial model preparations and intact cells. Cross-linking fixatives were phosphate-buffered solutions of 2 and 4% paraformaldehyde (pH 7.3), 4% paraformaldehyde with 0.1% glutaraldehyde, Zamboni's fixative, and Bouin's fixative. Fixation was performed for 1 hr at 4C. The coagulating fixatives were methanol, ethanol, and acetone with an incubation time of 30 min at −20C.
Immunocytochemistry
Artificial Models and Cultured Cells. Artificial model preparations were made of P16-coupled Sepharose beads that were air-dried on object slides. For intact cell preparations, cells were grown on 8-well chamber slides until 80% confluency. After washing with PBS, slides were incubated with the appropriate fixative. After washing, cells were permeabilized with methanol, ethanol, or acetone series (100, 50, 25%) each step 15 min at −20C. To decrease aspecific binding of antibodies slides were acetylated using a triethanolamine solution (0.1 M triethanolamine, pH 8.8, acetic acid anhydride 0.25%) (Hayashi et al. 1978). Immunoreactivity of the MP16-2 antibody was visualized using indirect immunoperoxidase or immunofluorescence methods. Secondary antibodies were rat anti-mouse immunoglobulins conjugated with horseradish peroxidase (RαM-PO; Dako) or rat anti-mouse immunoglobulins conjugated with fluorescein di-isothiocyanate (RαM-FITC; Dako). Primary as well as secondary antibodies were diluted in PBS containing 2% BSA and 0.1% Triton X-100. Specificity was investigated by preabsorbing MP16-2 antibody solution with various concentrations (0, 0.1, 0.5, 1, 10 μg/ml) of P16-coupled thyroglobulin for 30 min at RT. Immunoglobulin type-matched mouse MAbs against S-100 (Mα-S100; IgG2a) and follicle-stimulating hormone (Mα-FSH;IgG1) (Biogenex Laboratories; San Ramon, CA) or dilution buffer were used as negative controls. Slides were incubated with primary (diluted 1:100) or secondary antibodies (diluted 1:300) for 90 min at RT and subsequently washed with PBS. Peroxidase was visualized using 0.06% diaminobenzidine and 0.015% H2O2 in PBS. Object slides incubated with RαM-FITC were mounted in a mixture of glycerol (Merck) 90%, Tris-HCl (Merck) 10%, NaN3 (Merck) 0.1%. All other slides were mounted in Permount (Fisher; Fair Lawn, NJ). Slides mounted in Permount were stored at RT. Slides used for immunofluorescence were stored in the dark at 4C.
Staining results were examined using a Zeiss Axiophot microscope (Carl Zeiss; Oberkochen, Germany). Confocal laser scanning microscopy was carried out using a Nikon Diaphot (Nikon; Tokyo, Japan) and a Bio-Rad MRC 1000 (Bio-Rad; Hemel Hampstead, UK) equipped with a krypton-argon laser as described previously (Scheenen et al. 1996).
Tissues. Adult male rats (Wistar) were treated with testosterone by sc implantation of a timed-release pellet containing 50 mg testosterone (Innovative Research of America; Sarasota, FL). After 72 hr the rats were sacrificed by cervical dislocation and adrenals, colon, kidney, liver, pancreas, prostate, small intestine, and spleen were removed. In a separate experiment, pancreas and intestinal tissues of 1-, 7-, 14-, 21-, 28-, and 100-day-old rats were obtained. Specimens of the tissues were fixed overnight at 4C in 4% paraformaldehyde or Bouin's fixative and paraffin-embedded. Sections 4 μm thick were mounted on object slides, de-waxed, rehydrated through a series of ethanol solutions, and treated with 3% H2O2 in methanol for 3 min to block endogenous peroxidase activity. For heat-induced antigen retrieval (Cattoretti et al. 1993), the slides were boiled in 0.01 M sodium citrate, pH 6.0, for 15 min in a microwave oven at 800 W.
Slides were incubated with MP16-2 antibody (1 mg/ml) diluted 1:100 in antibody buffer (PBS containing 1% BSA, 0.1% Tween-20, and 0.01% SDS) for 18–24 hr at 4C. Specificity of the staining method was investigated by preabsorbing MP16-2 antibodies with various concentrations (0, 0.1, 0.5, 1, 10 μg/ml) of P16-coupled thyroglobulin for 30 min at RT. Bound antibodies were visualized with ABC Elite Vectastain reagents (Vector Laboratories; Burlingame, CA) using H2O2 and diaminobenzidine as chromogen. Sections were placed in a developing medium containing 0.006% H2O2 and 1 mg/ml diaminobenzidine and incubated for 10 min at RT.
Results
Characterization of the Antibody
Western blotting analysis was used to evaluate the immunoreactivity of MAb MP16-2 towards ODC in different cell and tissue preparations. The antibody reacted with a 53-kD protein of the same molecular weight (Mr) as ODC (Figure 1, Lanes 1-3). ODC-transfected NIH 3T3 cells expressed high levels of ODC protein (Figure 1, Lane 1) compared to nontransfected cells, which showed only a very weak band (not shown). In mouse kidney cytosol, other weaker bands with higher Mr are present, which may represent complexed forms of ODC (Figure 1, Lane 2). The antibody did not react with purified ODC complexed with [3H]-DFMO (Figure 1, Lane 4), indicating that binding of DFMO masks or changes the epitope recognized by MP16-2. ELISA studies showed that the MAb MP16-2 detected ODC in cell and tissue homogenates adhered to plastic surfaces (Figures 2B and 2C). One microgram of antibody was able to detect 60 μg of cell homogenate and 6.5 ng of ODC purified from stimulated mouse kidney, which corresponded to 3.5 pU and 330 pU of ODC activity, respectively. For comparison, Figure 2A shows the ELISA standard titration curve for P16 peptide conjugate of thyroglobulin.
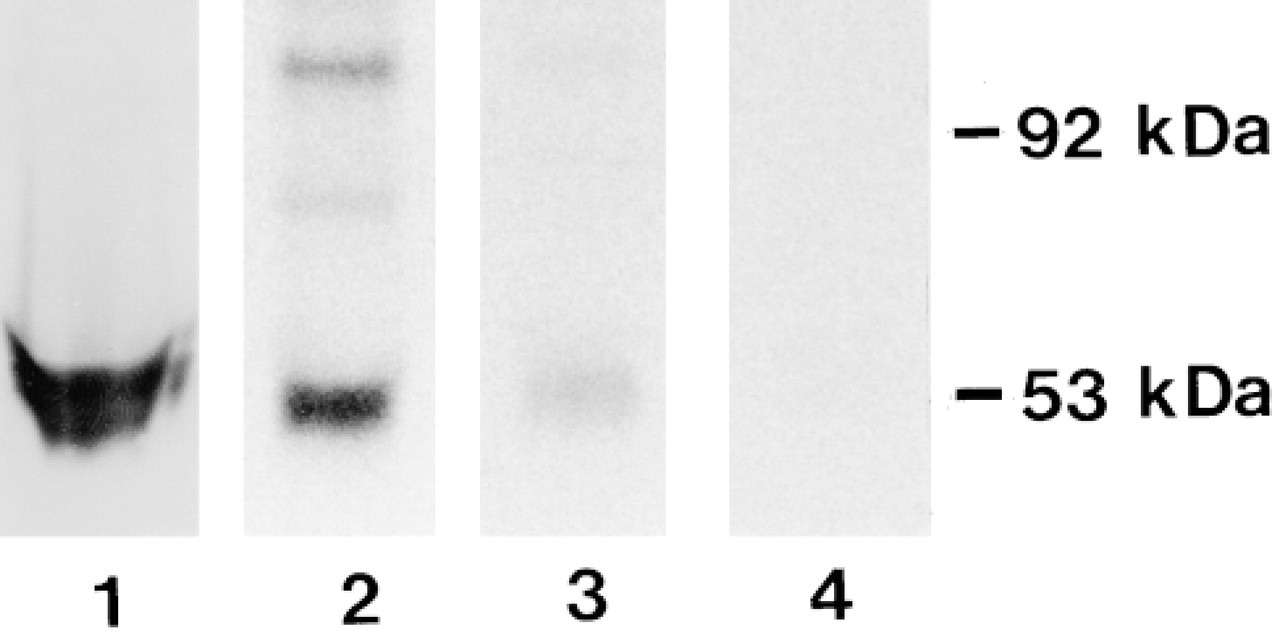
Western blotting of different ODC preparations of a cell homogenate (Lane 1) and testosterone-stimulated mouse kidneys (Lanes 2-4) using the MP16-2 antibody. Lane 1, ODC-overexpressing NIH 3T3 cells; Lane 2, kidney cytosol; Lane 3, purified ODC; Lane 4, purified ODC labeled with [3H]-DFMO.
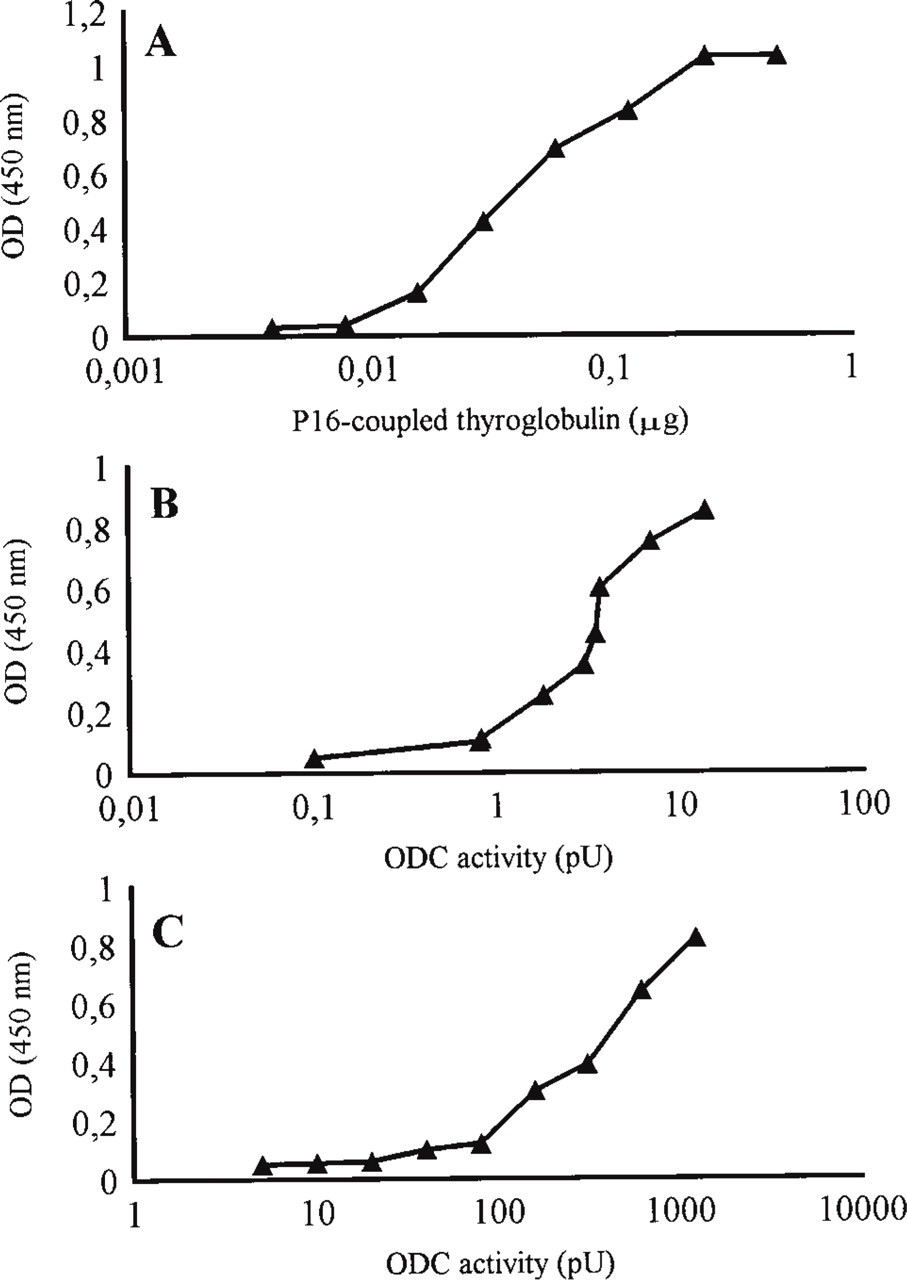
Typical ELISA curves of MAb MP16-2. Wells were coated with dilutions of (A) P16 peptide-conjugated thyroglobulin (B) cell homogenate of HCT 116 cells, or (C) ODC purified from kidney cytosol. Results are means of two measurements performed in two separate experiments.
Methodological Studies
The effects of different fixatives on MP16-2 immunostaining in artificial model preparations (DASS system) and the WISH cells are shown in Table 1. All crosslinking fixatives used were capable of retaining antigenicity of P16 in the DASS system (Figure 3), whereas the use of coagulating fixatives did not result in any immunostaining with MP16-2. Comparable results were obtained in experiments with cultured WISH cells. Fixation with 4% paraformaldehyde or Bouin's fixative provided the best results in intact cells.
The effects of fixation time, permeabilization procedures, and addition of glutaraldehyde in various concentrations to the 4% paraformaldehyde-containing fixative on immunostaining of P16-coupled Sepharose beads or WISH cells are shown in Table 2. The effects on intact beads or paraffin sections of beads were similar. In cultured cells, optimal staining results were achieved after 1 hr of fixation. After prolonged fixation times, staining intensity decreased. Treatment with a methanol series proved to be the best procedure to permeabilize cells, whereas all permeabilization methods tested did not affect immunostaining of the artificial model preparations. Glutaraldehyde concentrations higher than 0.1% decreased staining intensity in artificial model preparations as well as cultured cells (not shown).
Beads were not stained after incubation with control antibodies instead of the MP16-2 antibody (Figure 3, inset), whereas cultured cells showed nonspecific background staining. However, treatment of the cells with an acetylating solution as described by Hayashi et al. (1978) prevented this background staining.
Because fixation with 4% paraformaldehyde or Bouin's fixative provided the best results in the model studies, these fixatives were applied to tissue sections. ODC immunoreactivity was best preserved in rat tissues with fixation according to Bouin. Addition of 0.01% SDS to the antibody buffer solution increased the staining intensity considerably. Further increase in staining intensity and reduction of background staining were achieved with the antigen retrieval procedure using citrate buffer.
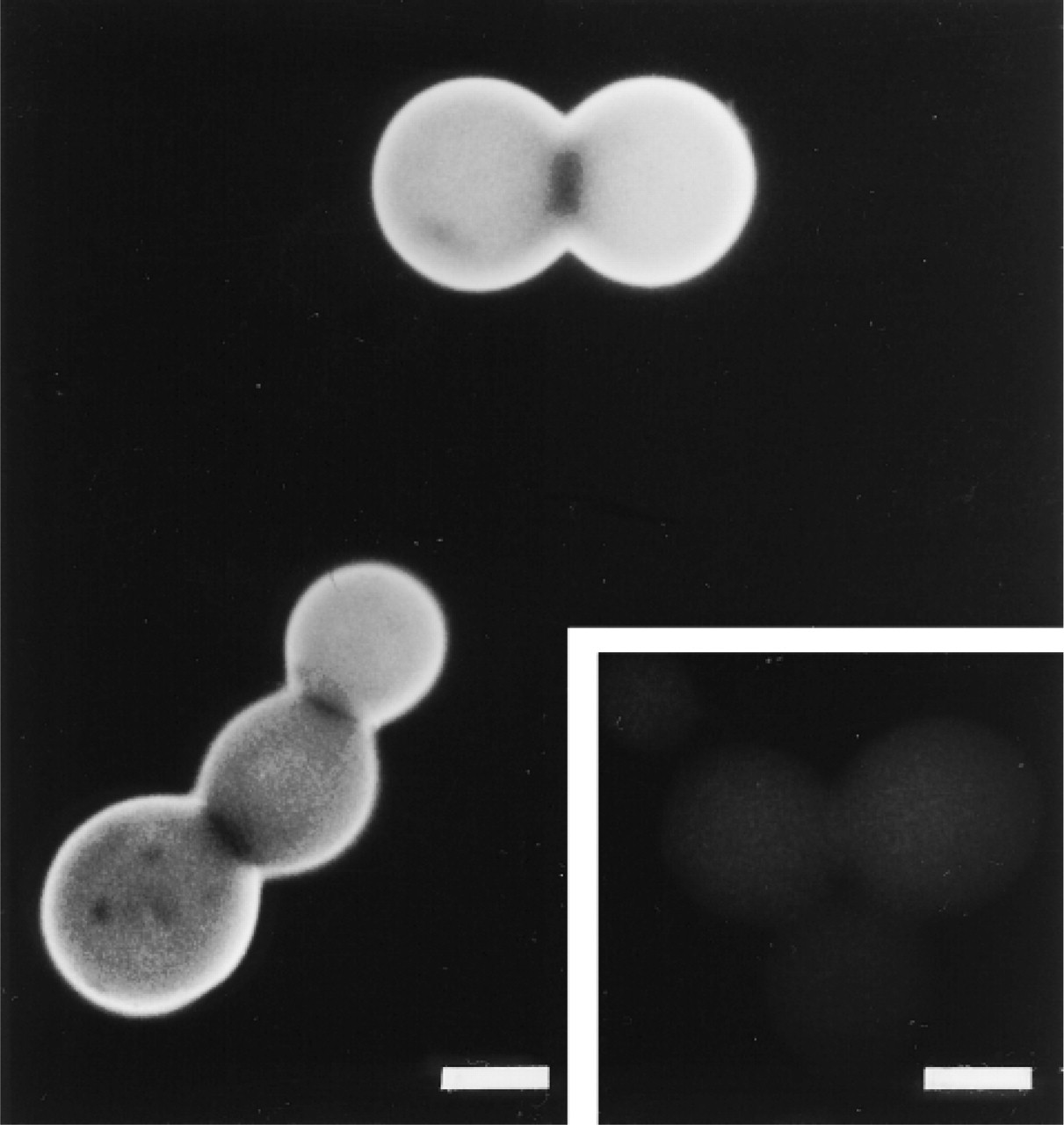
Photomicrograph of P16 peptide-coupled Sepharose beads after paraformaldehyde fixation and staining with MAb MP16-2 using the indirect immunofluorescence technique. (Inset) Control staining with irrelevant MAb Mα-FSH. Staining was absent after using MAb MP16-2 when the beads were fixed with coagulating fixatives. Bars = 50 μm.
Effect of fixation methods on MP16-2 immunostaining a
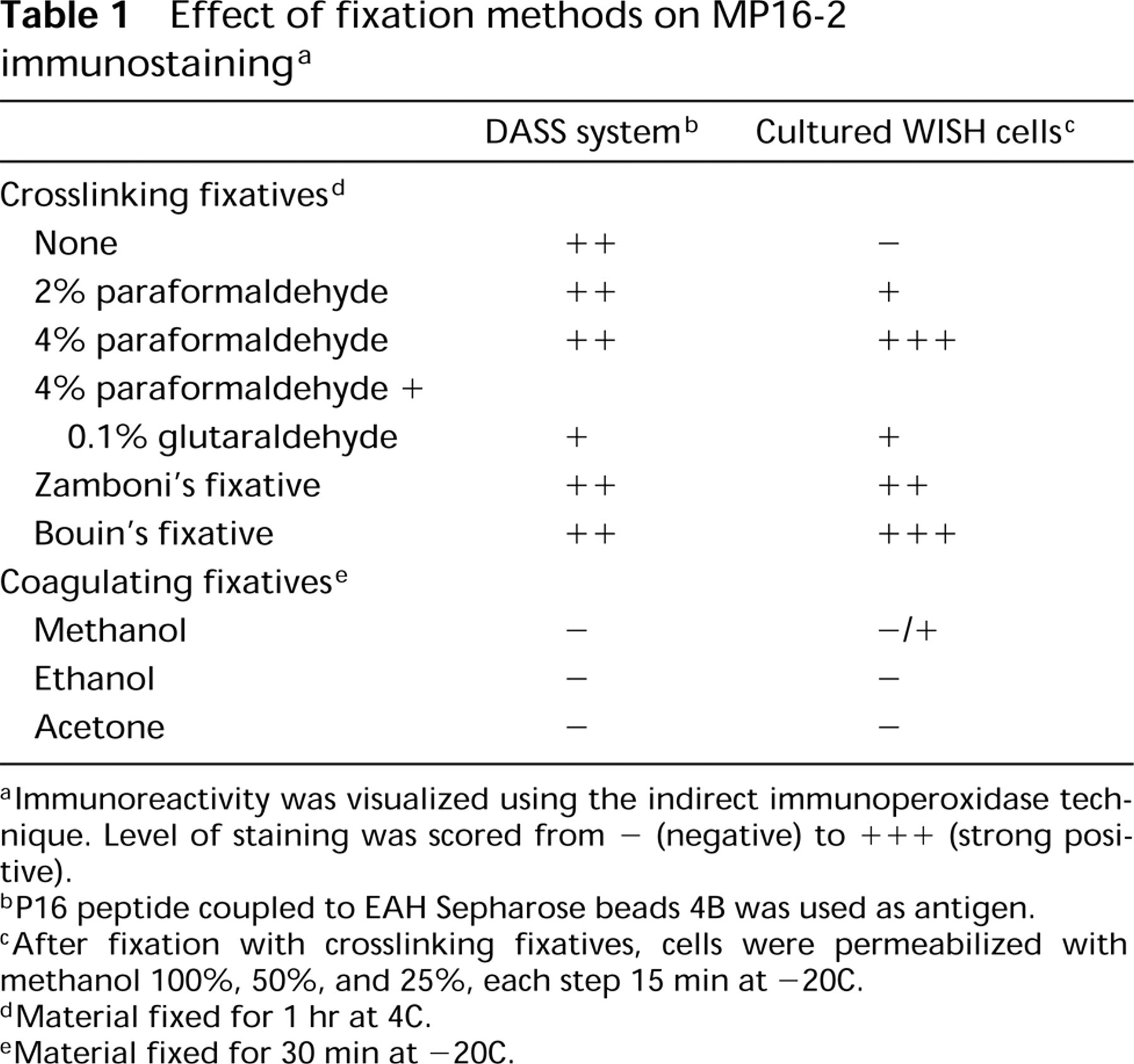
aImmunoreactivity was visualized using the indirect immunoperoxidase technique. Level of staining was scored from - (negative) to + + + (strong positive).
bP16 peptide coupled to EAH Sepharose beads 4B was used as antigen.
cAfter fixation with crosslinking fixatives, cells were permeabilized with methanol 100%, 50%, and 25%, each step 15 min at −20C.
dMaterial fixed for 1 hr at 4C.
eMaterial fixed for 30 min at −20C.
Effect of fixation time, permeabilization procedures, and glutaraldehyde on MP16-2 immunostaining a
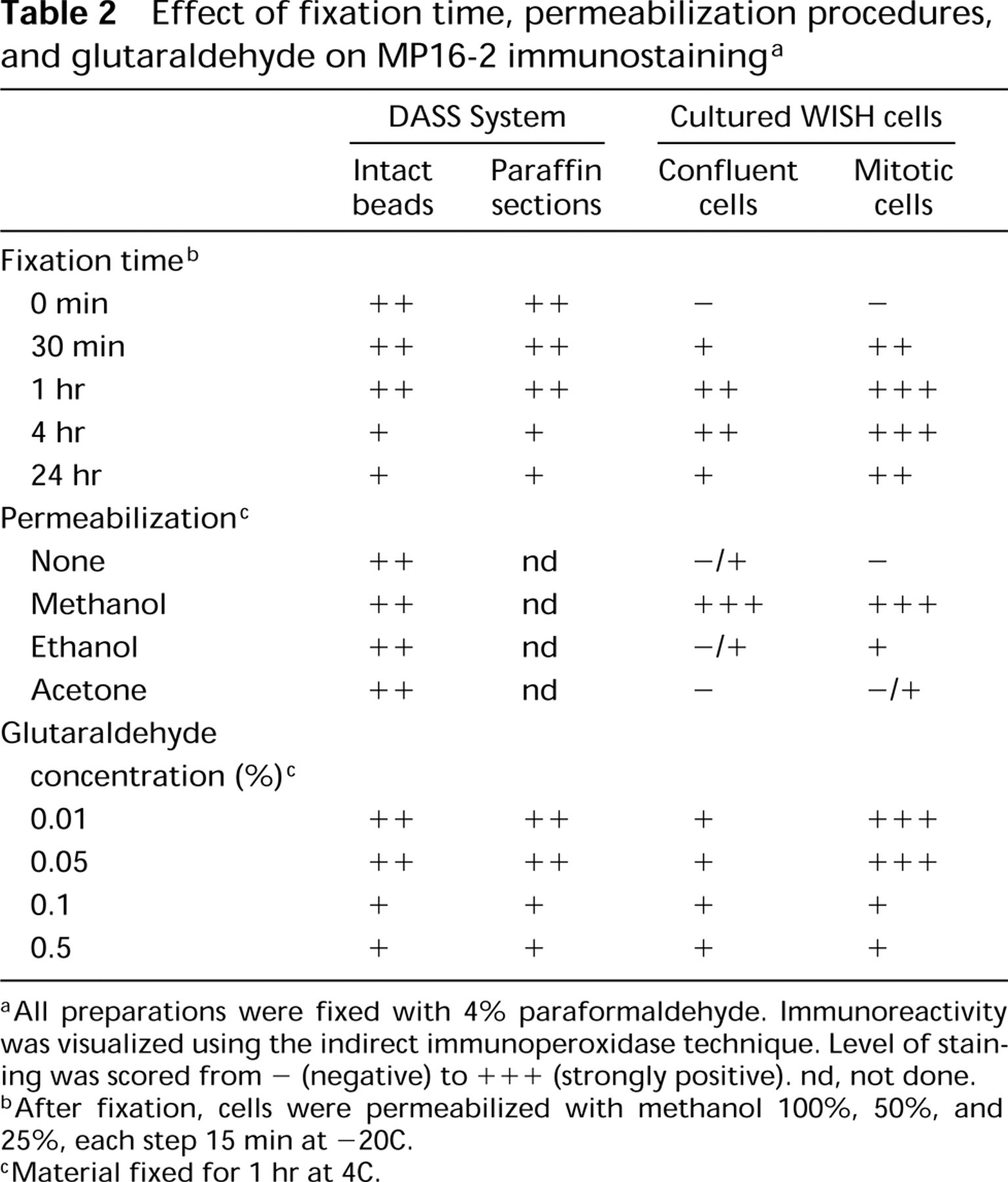
aAll preparations were fixed with 4% paraformaldehyde. Immunoreactivity was visualized using the indirect immunoperoxidase technique. Level of staining was scored from - (negative) to +++ (strongly positive). nd, not done.
bAfter fixation, cells were permeabilized with methanol 100%, 50%, and 25%, each step 15 min at −20C.
cMaterial fixed for 1 hr at 4C.
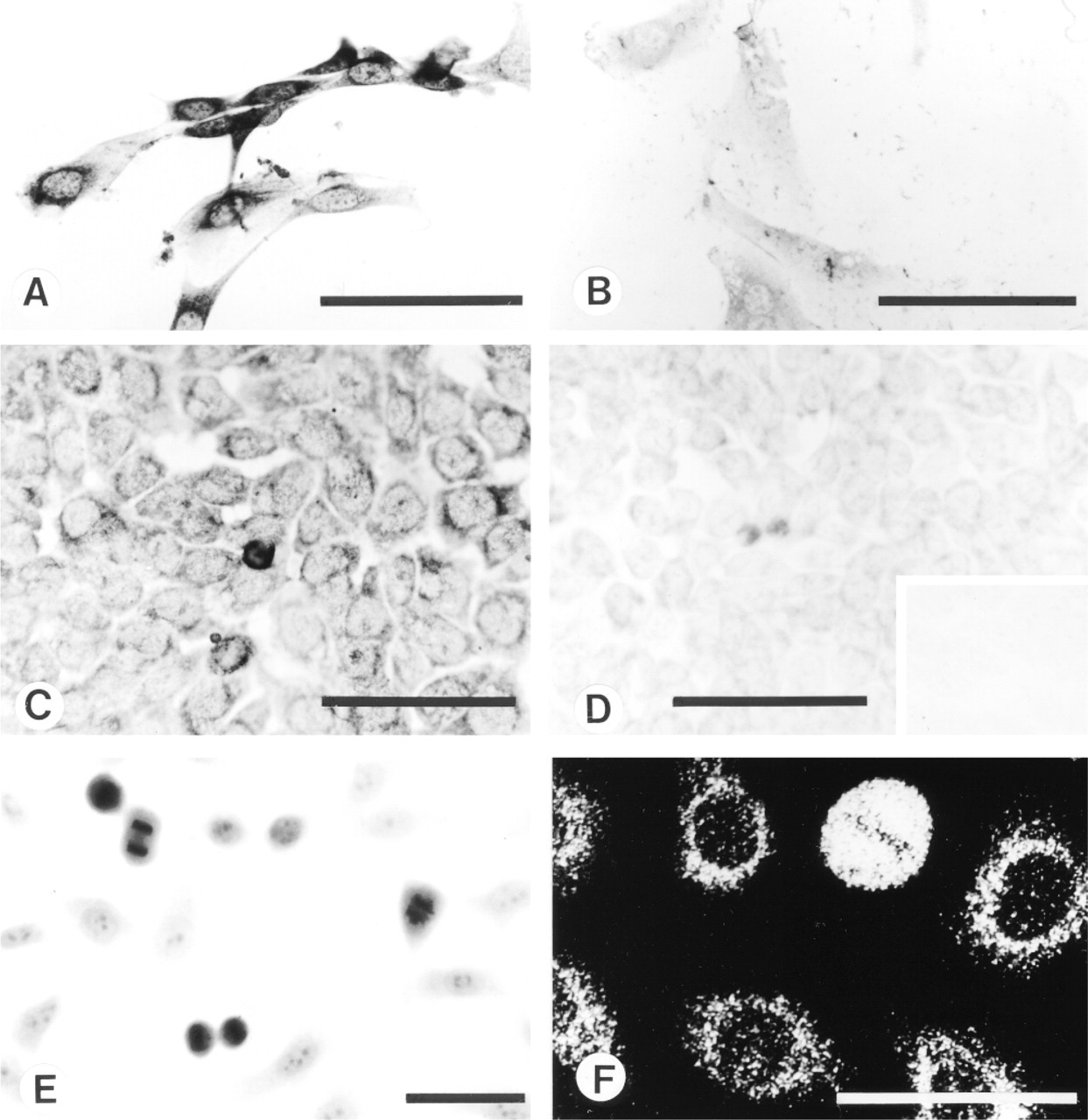
Cultured cells stained with MAb MP16-2 using indirect immunoperoxidase (A-E) or indirect immunofluorescence (F) technique. Staining of ODC-overexpressing (A) and normal (B) NIH 3T3 cells. Staining of HCT 116 cells after preabsorption with 0.1 (C), 0.5 (D) or 1 (D, inset) μg/ml of P16 peptide-conjugated thyroglobulin. Staining of human amnion WISH cells using light (E) or confocal laser scanning (F) microscopy. Note the strong nucleoplasmic and moderate, predominantly perinuclear staining of cultured mitotic and confluent cells, respectively. Bars = 50 μm.
MP16-2 immunostaining studies of different cell lines showed that all cells were positive for ODC. Immunostaining was substantially increased in ODC-transfected cells (Figure 4A) compared with nontransfected cells (Figure 4B). Mitotic cells were more intensely stained than confluent cells (Figures 4C-4F). In confluent cells, ODC staining was predominantly cytoplasmic, particularly in the perinuclear region, as revealed by confocal scanning microscopy (Figure 4F).
Distribution patterns of ODC immunoreactivity in rat tissues are summarized in Table 3 and Figure 5. In testosterone-treated rats, strong cytoplasmic staining was found in proximal tubules of kidney (Figure 5C), acinar cells of prostate, and adrenal medullary cells. In the small intestine of 7-, 14-, and 21-day-old rats, immunoreactivity was present in both crypts and villi, whereas in 28-day-old and adult (100-day-old) rats (Figure 5A) strong positivity for ODC was found in crypts only. In the pancreas of neonatal (1-day-old) rats, islets of Langerhans showed strong cytoplasmic staining (Figure 5B). In the pancreas of 14-day-old rats, heterogeneous staining of exocrine cells was observed (Romain et al. 1998).
Localization pattern of ODC obtained with immunostaining in rat tissues a
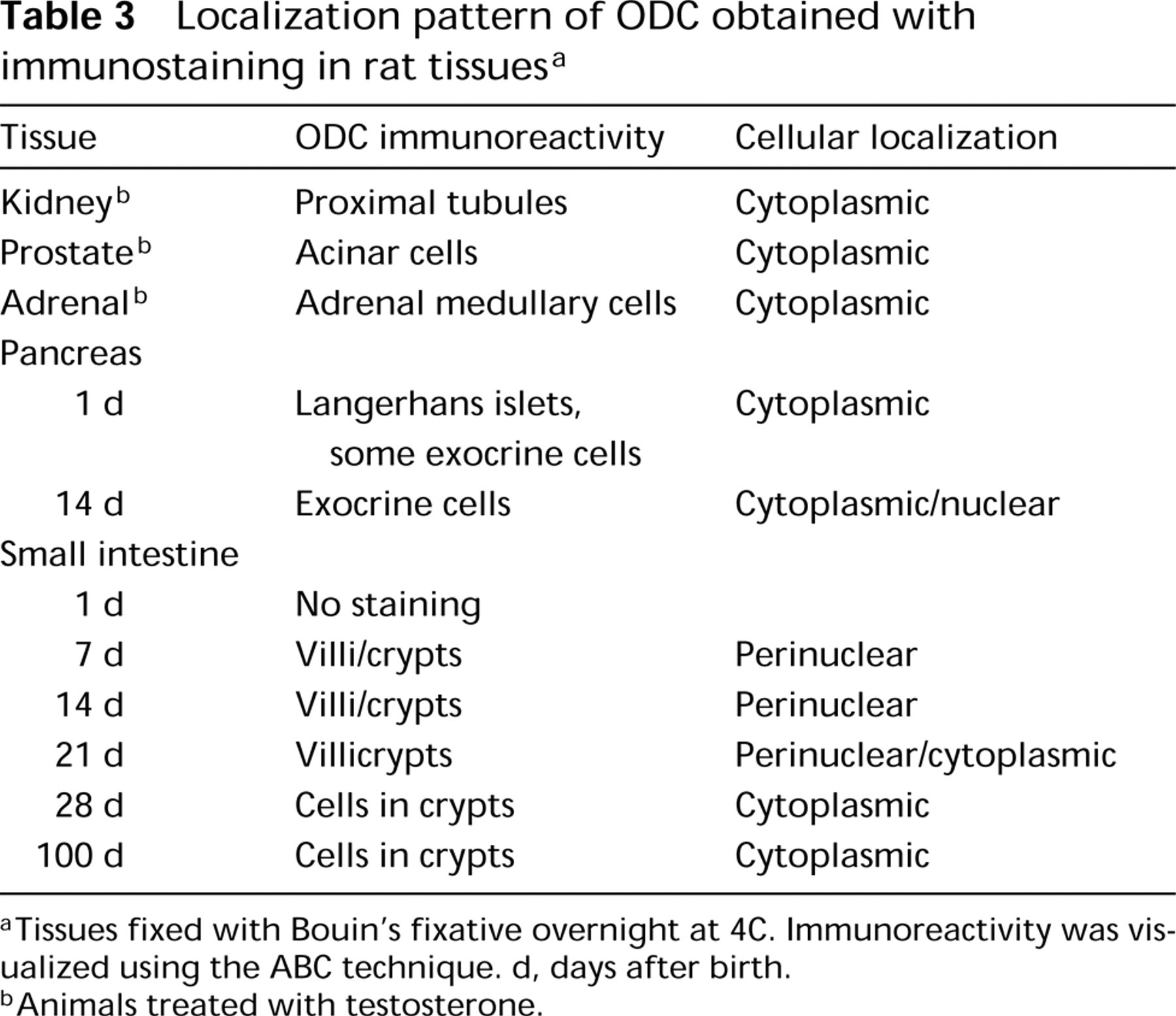
aTissues fixed with Bouin's fixative overnight at 4C. Immunoreactivity was visualized using the ABC technique. d, days after birth.
bAnimals treated with testosterone.
Preabsorption with 1 μg/ml of P16 peptide or more completely eliminated staining of cultured cells and all tissues investigated (Figures 4C, 4D, and 4D inset). No distinct differences were observed between results obtained with immunoperoxidase and immunofluorescence visualization methods.
Discussion
The purpose of this study was to systematically develop and optimize a reliable method for in situ detection of ODC utilizing an MAb (MP16-2) directed against an artificial antigen representing an ODC epitope (P16; amino acid residues 345-360). Authenticity of the P16 ODC epitope was proved by comparing the selected sequence with the primary sequences of other proteins. The sequence -WGPTC-, recognized by MP16-2, is present in all known eukaryote ODCs (Schipper et al. 1993). Immunoblotting results of the present study further confirm that the MP16-2 antibody also reacts with the epitope in the native ODC. Interestingly, the antibody did not bind to ODC complexed with [3H]-DFMO, which is in agreement with studies by Poulin et al. (1992) and Tobias and Kahana (1993). These authors reported that the amino acid Cys360, present in our selected epitope, is essential for ODC activity. Because DFMO-labeled ODC was not recognized by MP16-2, DFMO-treated cells and tissues could have been used for additional negative controls in the ODC cytochemical staining. However, the use of DFMO in vivo has significant drawbacks. Because ODC has a very short half-life, large and frequent doses of DFMO would be needed. Furthermore, the inhibitor is not taken up well by cells and it has to compete with ornithine to bind to the active site of ODC, which further complicates inhibition. Finally, it is also not certain whether different cell types or sub-cellular compartments take up DFMO in a similar way. Therefore, we did not perform these control studies.
Our ELISA experiments showed that the MP16-2 antibody could also be used to detect ODC in homogenates of cells or tissues coated to plastic surfaces. The difference in calculated sensitivity between the different ODC preparations (purified ODC, cell homogenate) might be due to variations in the amount of ODC epitope that became attached to the plastic surfaces.
In contrast to immunochemical procedures, e.g., ELISA, immunocytochemical staining procedures take place in a far more chemically complex matrix. Model systems using artificial matrices have proved to be valuable tools to study the complexity of (immuno)cytochemical processes (Van der Ploeg and Duijndam 1986). By gradually increasing the complexity of the models studied (artificial models, cultured cells, tissues) the reliability of immunocytochemical staining procedures can be analyzed and improved. To study the preservation of the ODC epitope in immunocy-tochemical processes, we used the DASS system as artificial model (Capel 1975; Streefkerk et al. 1975). This model has the advantage that antigens are covalently bound to Sepharose beads. These beads have microscopic dimensions and can easily be manipulated to imitate actual conditions in cells and tissues (Van der Ploeg and Duijndam 1986). Results with the DASS system and cultured cells showed that P16/ODC anti-genicity is sufficiently preserved in beads and cells fixed with crosslinking fixatives.
In contrast, if coagulating fixatives were used the intensity of immunostaining diminished. A frequently occurring problem with coagulative fixatives is that antigens are precipitated and extracted from biological matrices (Larsson 1993; Polak and Van Noorden 1997). Because in our DASS experiments the antigens are covalently immobilized to the beads, we assume that the coagulant fixatives may have altered or destroyed the P16/ODC epitope structure, preventing binding of the MP16-2 antibody. Interestingly, the epitope remains preserved if, after fixation with cross-linking fixatives, cells are exposed to coagulating fluids used for permeabilization of cells.
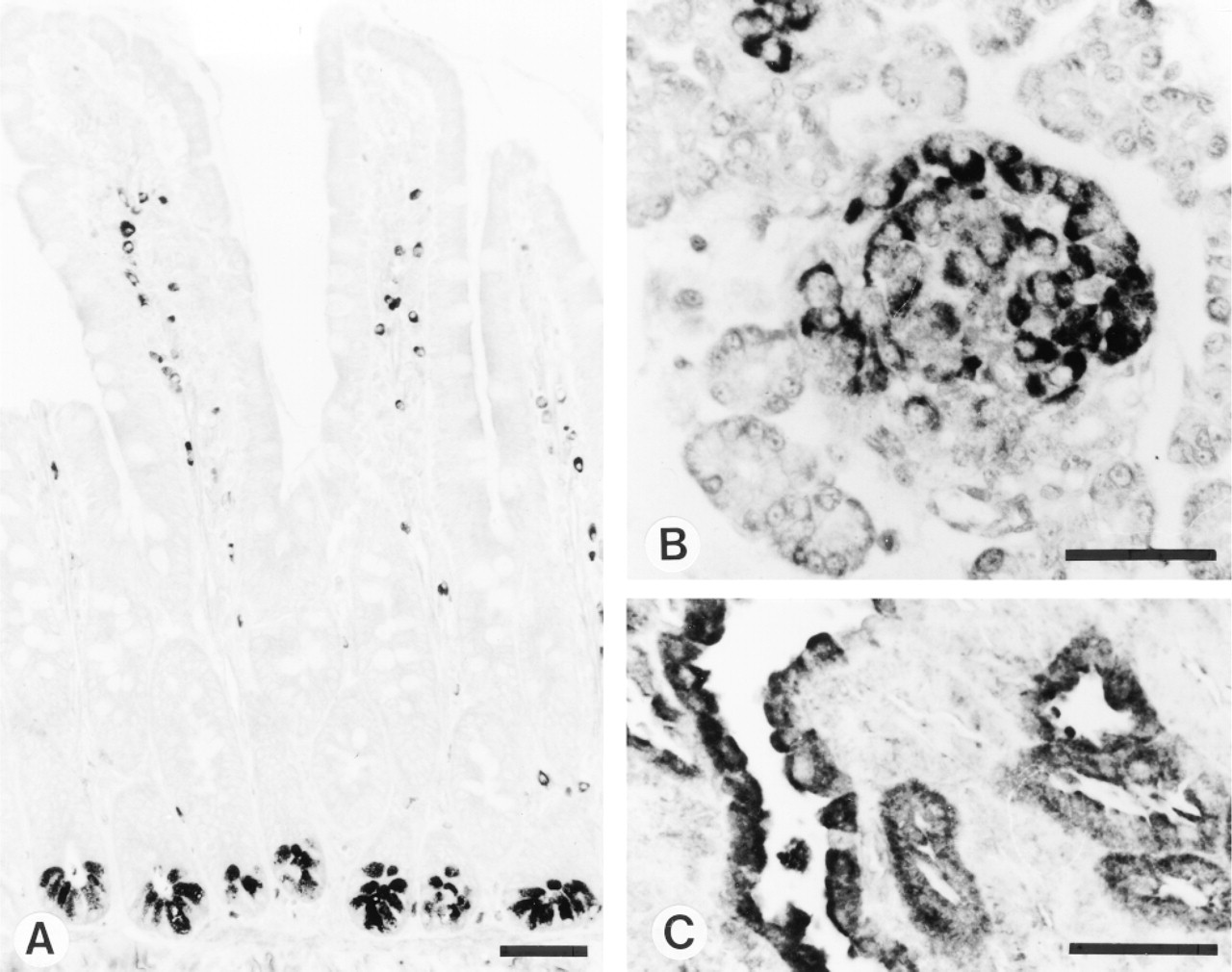
Indirect immunoperoxidase staining of rat tissues with MAb MP16-2. (A) Small intestine of adult rat. (B) Pancreas of neonatal rat. (C) Kidney of testosterone-treated rat. Note the immunoreactivity in the intestinal crypts, pancreatic islets, and renal proximal tubules, respectively. Bars = 50 μm.
Intact cells have to be made permeable to antibodies, especially after fixation with crosslinking fixatives that are known to create diffusion barriers (Larsson 1993). The results of our study confirm these findings, i.e., if permeabilization was not carried out after the use of a crosslinking fixative the staining pattern was completely different. Acetone, ethanol, and methanol are widely used to permeabilize cells for flow cytometric studies and are therefore also applicable to permeabilization of cells for immunocytochemical studies (see, e.g., Cattaneo et al. 1983). To permeabilize cells these substances were tested. Methanol was the only alcohol that provided good permeabilization of the cultured WISH cells.
The preservation of the epitope by fixatives containing both 4% formaldehyde and 0.05% glutaraldehyde opens perspectives for the application of MP16-2 in electron microscopic studies.
On the basis of the results obtained in the model studies, we tested the MP16-2 antibody on rat tissues fixed with 4% paraformaldehyde or Bouin's fixative. Initial experiments resulted in rather low and poorly reproducible immunostaining of the tissues. With the addition of SDS to the antibody-containing solution, staining was dramatically increased. This observation is compatible with reports from the literature showing that immunogenicity of a number of antigens is retrieved after SDS treatment of cell and tissues (Brown et al. 1996). Citrate buffer treatment, another antigen retrieval step, further improved the intensity of ODC staining and decreased background staining. A possible hazard of this heat-induced antigen retrieval step is the increased prominence of plasma cell immunoglobulins or endogenous biotin that can react with the secondary antibody or the ABC reagent, respectively. Therefore, the stained cells that were observed in the lamina propria of intestinal villi could be due to nonspecific staining of plasma cells. Nonspecific staining due to endogenous biotin is not likely in our studies because the use of the indirect immunoperoxidase method without the streptavidin-biotin reagent gave similar localization patterns (data not shown).
These observations illustrate the increasing complexity of biological matrices in tissues compared to single cells and artificial models. Antigens in tissues, although well preserved after fixation, might be masked or linked to other components (e.g., by hydrogen or disulfide bonds) and therefore hardly accessible to antibodies.
Although the present study was not primarily meant to investigate the intracellular localization of ODC in cultured cells, interesting results were obtained. Under optimal conditions, ODC showed a similar localization pattern in all tested cell lines. In resting cells, ODC staining was moderate and predominantly in the perinuclear region, whereas mitotic cells were strongly and homogeneously stained. Previous studies on stimulated murine macrophage-like RAW 264 cells (Greenfield et al. 1986) and ODC-overproducing CHO cells (Anehus et al. 1984) reported that ODC was localized exclusively or predominantly in the cytoplasm of cells. However, in a murine cell line of epidermal origin (JB6), immunoreactive ODC protein was found both in the cytoplasm and the nucleoplasm but not in nucleoli (Greenfield et al. 1986). In growing cell fractions of human keratinocytes, ODC showed a peri-nuclear and nuclear distribution pattern. When cell growth was arrested, ODC distribution became more diffuse and cytoplasmic (Pomidor et al. 1995).
The optimized immunocytochemical method was also applied to various rat tissues. In testosterone-treated rats, intense staining of ODC was detected in proximal tubules of kidney and acinar cells of prostate, which is in agreement with previous biochemical, enzyme cytochemical, and hybridocytochemical studies (Persson et al. 1984; Zagon et al. 1984; Dorn et al. 1985; Blackshear et al. 1989; Koibuchi et al. 1993). Testosterone treatment did not change the intensity of staining in other tissues tested, e.g., small intestine and pancreas (not shown).
ODC expression has been shown to change during growth and maturation of developing tissues. In developing rat pancreas and small intestine, we observed age-related localization patterns that might reflect the changes of ODC expression during growth and maturation of these organs (manuscript in preparation).
In conclusion, this report describes conditions required for an optimal detection of ODC with the MP16-2 monoclonal antibody in homogenates, intact cells, and tissue sections. Using optimized techniques, we demonstrated high ODC immunoreactivity in cells and tissues that are engaged in growth or differentiation processes. Moreover, the ELISA technique provides a new and valuable quantitative assay for ODC protein in homogenates of cells and tissues.
Footnotes
Acknowledgements
Supported by the Dutch Cancer Society (grant NKB-93-599) and the Nijbakker Morra Foundation.
We gratefully acknowledge the help of Dr Martin Sauerbeck and Prof Dr Peter Bohley (Department of Physiological Chemistry, University of Tubingen, Germany) in the preparation of purified ODC. We thank Rob Rutten, Xander de Haan, Birgitte Walgreen, and Wilma Van Staveren for technical assistance. We are also grateful to Prof Dr Ron Van Noorden and Dr Wilma Frederiks (Department of Cell Biology and Histology, University of Amsterdam) for valuable comments and suggestions for this manuscript.