
Abstract
DLK/MUK/ZPK is a serine/threonine kinase that belongs to the mixed-lineage (MLK) subfamily of protein kinases. As is the case for most members of this family, relatively little is known about the physiological role of DLK/MUK/ZPK in mammalian cells. Because analysis of subcellular distribution may provide important clues concerning the potential in vivo function of a protein, an antiserum was generated against the amino terminal region of murine DLK/MUK/ZPK and used for localization studies in wild-type NIH 3T3 cells. Light microscopic immunocytochemistry experiments performed with the antiserum revealed that DLK/MUK/ZPK was specifically localized in a juxtanuclear structure characteristic of the Golgi complex. In support of this, treatment of cells with brefeldin A, a drug known to disintegrate the Golgi apparatus, caused disruption of DLK/MUK/ZPK peri-nuclear staining. Ultrastructural observation of NIH 3T3 cells also confirmed this localization, showing that most of the immunoreactivity was detected on membranes of the stacked Golgi cisternae. Consistent with localization studies, biochemical analyses revealed that DLK/MUK/ZPK was predominantly associated with Golgi membranes on fractionation of cellular extracts and was entirely partitioned into the aqueous phase when membranes were subjected to Triton X-114 extraction. On the basis of these findings, we suggest that DLK/MUK/ZPK is a peripheral membrane protein tightly associated with the cytoplasmic face of the Golgi apparatus.
O
Among the different MAPKKKs identified thus far and implicated in the regulation of the MAPK signaling pathways are members of the mixed-lineage kinase (MLK) family (Fanger et al. 1997). The MLKs constitute a recently identified subfamily of protein kinases which are characterized by a unique catalytic domain hybrid between those found in serine/threonine and tyrosine kinases. In addition, the MLKs typically contain in their extracatalytic regions amino acid sequence motifs that are believed to be involved in protein-protein interactions, such as an amino terminal src homology 3 (SH3) domain, two central leucine/isoleucine zipper motifs, and a carboxy terminal proline-rich region. To date, five distinct MLK family members have been characterized at the molecular level and four of them, designated MLK2/MST (Dorow et al. 1995; Katoh et al. 1995), MLK3/SPRK/PTK1 (Ezoe et al. 1994; Gallo et al. 1994; Ing et al. 1994), DLK/MUK/ZPK (Holzman et al. 1994; Reddy and Pleasure 1994; Hirai et al. 1996), and LZK (Sakuma et al. 1997), activate preferentially the JNK/SAPK pathway when overexpressed in transiently transfected cells (Fan et al. 1996; Hirai et al. 1996,1997; Rana et al. 1996; Teramoto et al. 1996; Tibbles et al. 1996; Sakuma et al. 1997). In fact, these kinases act as MAPKKK components of this signaling pathway by phosphorylating and activating SEK1/MKK4/JNKK, a direct upstream activator of the JNKs/SAPKs.
Despite their involvement in the JNK/SAPK cascade, relatively little is known about the mechanisms of regulation and the physiological roles of the different MLK family members in mammalian cells. A potential function for the DLK/MUK/ZPK gene in biological processes related to differentiation was recently postulated on the basis that its expression in embryonic and adult mouse tissues is restricted to highly specialized cell populations, such as neurons and epithelial cells (Blouin et al. 1996; Nadeau et al. 1997). To obtain further insights into the function of DLK/MUK/ZPK, a specific antiserum was raised against this protein kinase to carry out a detailed examination of its localization in NIH 3T3 cells. Immunocytochemical and biochemical analyses have demonstrated that DLK/MUK/ZPK is specifically localized to the Golgi complex, where it behaves like a peripheral membrane protein that faces the cytosol. Such a location suggests a role for this enzyme in the function of the Golgi apparatus.
Materials and Methods
Production of DLK/MUK/ZPK-specific Antisera
A portion of the mouse DLK/MUK/ZPK cDNA (nucleotides 42-711) (Blouin et al. 1996) was cloned into a pRSET expression vector (Invitrogen; Carlsbad, CA) and expressed in E. coli strain BL21 pLys for production of a recombinant protein containing a polyhistidine tag fused to the NH2 terminal 223 amino-acid residues of DLK/MUK/ZPK. The fusion protein was purified under denaturing conditions on an Ni2+ -NTA-Agarose column using the Xpress protein purification system (Invitrogen). Rabbits were immunized with 100 μg of purified recombinant DLK/MUK/ZPK protein emulsified in TiterMax (CytRx; Norcross, GA) adjuvant. Rabbits were bled 8 weeks after the immunization and one of these antisera was used for immunoblotting, immunoprecipitation, and immunocytochemistry experiments.
Cell Culture
All cell lines used in this work were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 25 μg/ml amphotericin B at 37C in a 5% CO2 atmosphere.
DLK/MUK/ZPK Expression in COS-1 Cells
The cDNA encoding mouse DLK/MUK/ZPK (Blouin et al. 1996) was inserted into the cytomegalovirus promoter-based eukaryotic expression vector pcDNA3 (Invitrogen) as a HindIII- NotI fragment. COS-1 cells (3 × 105) plated in 60-mm dishes were transfected with 5 μg of the expression construct using lipofectamine (Life Technologies; Rockville, MD) according to the manufacturer's protocol. After overnight incubation, the transfection mixture was removed and replaced with fresh medium. Immunoblot analyses were carried out 48 hr after transfection.
Preparation of Cell Lysates and Immunoblotting
Cells (control COS-1, DLK/MUK/ZPK-transfected COS-1, wild-type NIH 3T3 and Swiss 3T3, fibroblasts) were lysed for 30 min at 4C in PBS containing 1% Triton X-100, 10 mM NaPPi, 100 mM NaF, 0.2 mM Na3VO4, 0.2 mM PMSF, 2 μg/ml leupeptin, and 1 μg/ml aprotinin (PBS-T). Lysates were then centrifuged at 12,000 rpm for 15 min at 4C, and the concentration of total protein in the supernatant fraction was quantitated using the Bio-Rad protein assay (Bio-Rad Laboratories; Mississauga, Ontario, Canada). Proteins were separated on 10% SDS-PAGE under reducing conditions and transferred onto polyvinylidene difluoride (PVDF) membranes (Boehringer Mannheim; Laval, Quebec, Canada) using a semidry transfer apparatus (Amersham Pharmacia Biotech; Baie d'Urfé, Quebec, Canada). Membranes were blocked overnight at 4C in 20 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Tween-20 (TBS-T) containing 5% skim milk powder. Blots were then incubated with the primary antibody for 1 hr at room temperature and the immunoreactivity was detected by enhanced chemiluminescence, using as secondary antibody a horseradish peroxidase-linked anti-rabbit antibody (ECL Western blotting kit; Amersham Pharmacia Biotech).
Immunocytochemistry
Light and electron microscopic immunocytochemistry experiments were carried out as described previously (Brown and Farquhar 1989; Blouin et al. 1997). DLK/MUK/ZPK antiserum was used at a dilution of 1:400 for both light and electron microscopic observations. Alkaline phosphatase-conjugated anti-rabbit IgG (1:1000 dilution; Sigma-Aldrich, Oakville, Ontario, Canada) and horseradish peroxidase-conjugated anti-rabbit IgG (1:200 dilution; Sigma-Aldrich) were used as secondary antibodies for light and electron microscopic immunocytochemistry, respectively.
Subcellular Fractionation
Subcellular fractionation of NIH 3T3 cells was performed on self-forming Percoll gradient as described previously (Morand and Kent 1986). Briefly, cells were homogenized in ice-cold TS buffer (10 mM Tris-HCl, pH 7.4, 250 mM sucrose, 1 mM PMSF, 1 μg/ml leupeptin, and 1 μg/ml aprotinin), and treated with 75 U/ml DNase I (Amersham Pharmacia Biotech) for 30 min at 37C. The homogenate was then layered on top of a 13.5% Percoll gradient and spun in a Beckman 50 Ti rotor for 1 hr at 17,000 rpm. After centrifugation, 1-ml fractions were collected from the top of the tube. Equal amounts of proteins from each fraction were precipitated by the method of Wessel and Flügge (1984) and analyzed by immunoblotting using either DLK/MUK/ZPK antiserum (1:1000 dilution) or (α-mannosidase II (Man II) antiserum (1:2000 dilution; supplied by Dr. K.W. Moremen, University of Georgia). A rabbit antiserum directed against G-protein β-subunits (1:1000 dilution; Chemicon International, Temecula, CA) was also used in these experiments as a marker for the plasma membrane.
Extraction and Trypsin Treatment of Membranes
NIH 3T3 cells were homogenized in 250 mM sucrose, 10 mM Tris-HCl, pH 7.5, 2 mM EDTA, 1 mM PMSF, 2 μg/ml leupeptin, and 1 μg/ml aprotinin with a Dounce homogenizer, followed by centrifugation at 600 × g for 3 min. The supernatant from this centrifugation was then spun at 100,000 × g for 1 hr at 4C in a Beckman SW50.1 rotor, and the resulting pellet (crude membrane fraction) was resuspended in the homogenization buffer containing either 1 M NaCl, 1 M KCl, or 0.1 M Na2CO3, pH 11.5. After incubation on ice for 30 min, the samples were centrifuged again at 100,000 × g for 30 min to obtain supernatants and membranes pellets. Extraction of crude membrane fraction with Triton X-114 was carried out as described by Bordier (1981). The supernatants and pellets of different treatments, as well as aqueous and detergent phases of Triton X-114 extraction, were subjected to SDS-PAGE, folowed by immunoblotting with DLK/MUK/ZPK antiserum.
Crude membrane fractions prepared as described above were also resuspended in 10 mM Tris-HCl, pH 8.0, 150 mM NaCl, with or without 1% Triton X-100. After incubation on ice for 5 min, trypsin (0.1 μg/ml) was added and the digestion was allowed to proceed at 4C for 20 min. Then protease inhibitors (2 mM PMSF, 20 μg/ml leupeptin, 20 μg/ml aprotinin) were added to neutralize the trypsin and the membranes were diluted with an equal volume of digestion buffer containing either 1% or 2% Triton X-100. After a 30-min incubation at 4C, the samples were centrifuged at 12,000 rpm in a microfuge for 15 min. The supernatants of these incubations were then immunoprecipitated for 3 hr at 4C with constant rotation, using either DLK/MUK/ZPK anti serum (1:250 dilution) or Man II antiserum (1:500 dilution) and protein A-Agarose beads. At the end of the incubation period, the immunocomplexes were collected by centrifugation at 12,000 rpm for 30 sec and washed twice with digestion buffer containing 1% Triton X-100. The final pellet was resuspended in 2 × Laemmli sample buffer, heated to 95C for 5 min, and fractionated by SDS-PAGE. DLK/MUK/ ZPK and Man II were detected by immunoblotting.
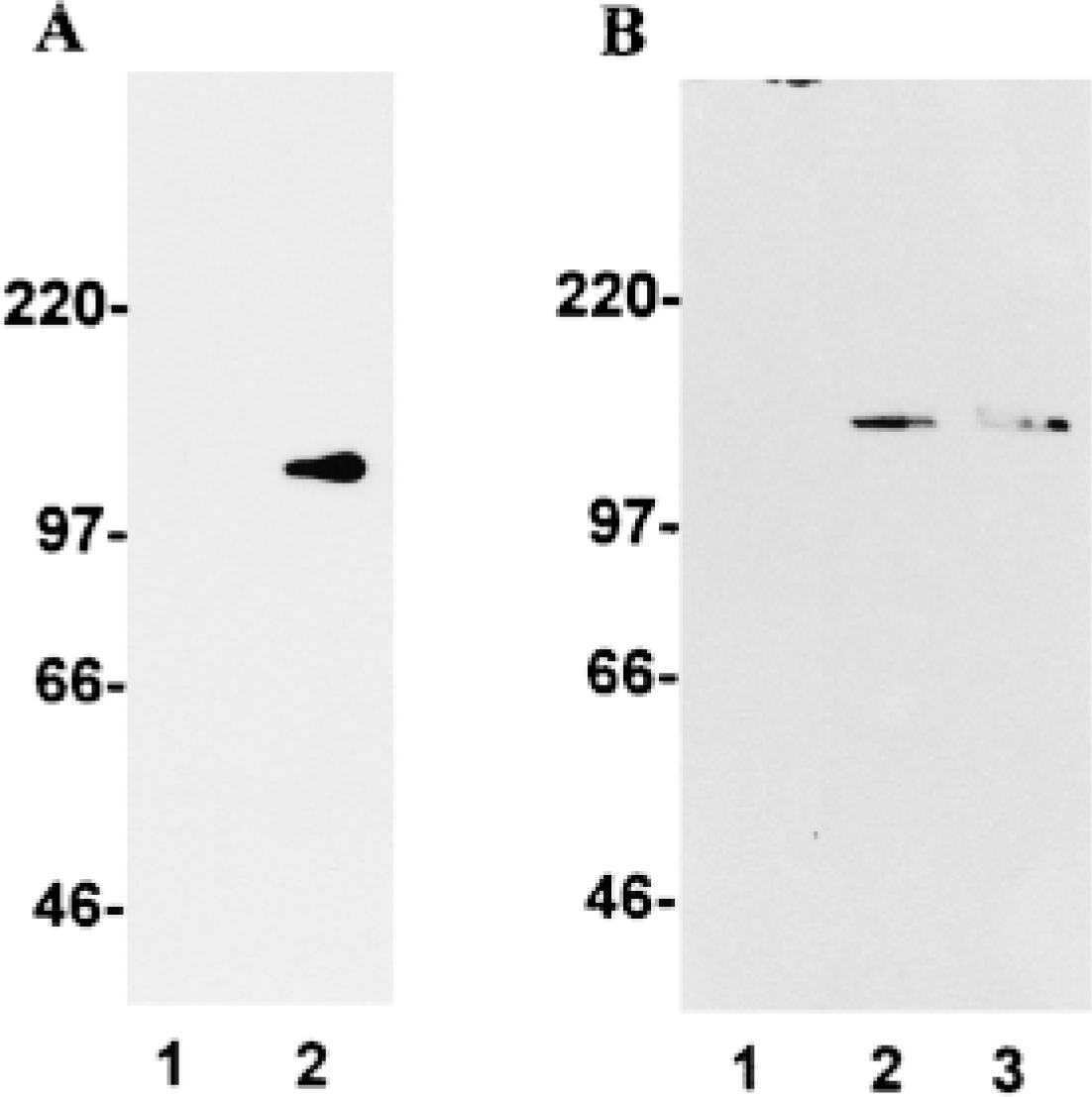
Specificity of DLK/MUK/ZPK antiserum. (A) Ten μg of cellular extracts prepared from control (Lane 1) and DLK/MUK/ZPK-transfected Lane 2) COS-1 cells was fractionated by SDS-PAGE and electrophoretically transferred to PVDF membranes. Blots were then incubated with DLK/MUK/ZPK antiserum and developed using enhanced chemiluminescence (ECL). (B) Twenty μg of cell lysates prepared from wild-type NIH 3T3 (Lanes 1 and 2) and Swiss 3T3 (Lane 3) cells was fractionated on SDS-PAGE and immunoblotted with either preimmmune serum (Lane 1) or DLK/MUK/ZPK antiserum (Lanes 2 and 3). Immunoreactive proteins were visualized by ECL.
Results
Characterization of Anti-DLK/MUK/ZPK Antisera
To investigate the biological function of the mixed-lineage kinase DLK/MUK/ZPK, we raised antisera in rabbits against a recombinant E. coli-produced fragment of the mouse DLK/MUK/ZPK protein. The specificity of one of these antisera was assayed by immunoblotting of cell lysates prepared from control and DLK/MUK/ZPK-transfected COS-1 cells and from wild-type NIH 3T3 and Swiss 3T3 fibroblasts. In immunoblot analysis carried out with lysates of COS-1 cells, the antiserum recognized a unique protein band of ∼130 kD only in DLK/MUK/ZPK transfectants (Figure 1A). The size of this protein corresponds to what has been previously reported for transfected mouse and rat DLK/MUK/ZPK (Holzman et al. 1994; Hirai et al. 1996). The antiserum also specifically detected a single band on lysates of wild-type NIH 3T3 and Swiss 3T3 cells that most likely represents endogenous DLK/MUK/ZPK (Figure 1B, Lanes 2 and 3). Endogenous DLK/MUK/ZPK in NIH 3T3 and Swiss 3T3 cells migrated as a slightly larger protein than the recombinant form obtained after transfection of COS-1 cells with the murine DLK/MUK/ZPK cDNA. No signal was detected tected in either cell type when equal amounts of rabbit preimmune serum were tested on cell extracts run in parallel (Figure 1B, Lane 1). These results demonstrated that DLK/MUK/ZPK antiserum was highly specific and therefore suitable for immunohistochemical localization of the DLK/MUK/ZPK protein.
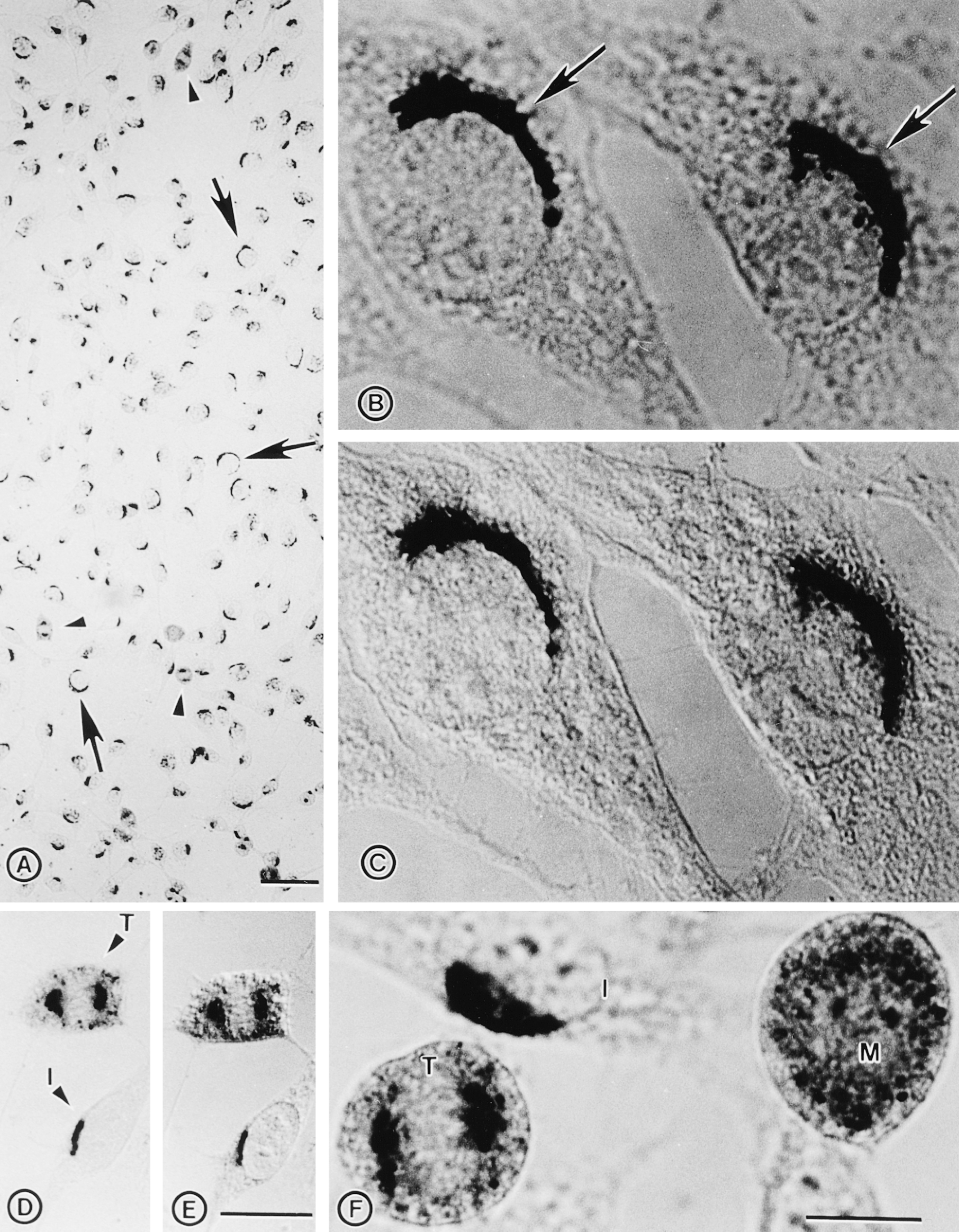
Immunocytochemical localization of endogenous DLK/MUK/ZPK in NIH 3T3 cells. NIH 3T3 cells grown on coverslips were fixed with methanol/acetone and stained with DLK/MUK/ZPK antiserum followed by alkaline phosphatase-conjugated secondary mouse anti-rabbit antibodies. (A) Staining of NIH 3T3 cells with DLK/MUK/ZPK antiserum revealed prominent localization of the signal in the Golgi region. Arrows and arrowheads mark interphase and mitotic cells, respectively. Bar = 50 μm. (B) High magnification of NIH 3T3 cells stained with antiserum to DLK/MUK/ZPK, showing preferential localization of the signal at a juxtanuclear site corresponding to the Golgi apparatus. Bar = 10 μm. (C) Same as B except that stained cells were observed under Nomarski interference microscopy. Bar = 10 μm. (D) DLK/MUK/ZPK exhibits a differential distribution in interphase (I) and telophase (T) cells. Bar = 20 μm. (E) Same as D except that stained cells were observed under Nomarski interference microscopy. Bar = 20 μm. (F) Hi5gh magnification of NIH 3T3 cells stained with antiserum to DLK/MUK/ZPK, showing differential localization of the signal in interphase (I), metaphase (M), and telophase (T) cells. Bar = 10 μm.
Localization of DLK/MUK/ZPK in Wild-type NIH 3T3 Cells
Subcellular localization of DLK/MUK/ZPK was initially examined in NIH 3T3 cells by immunohistochemistry with DLK/MUK/ZPK antiserum and alkaline phosphatase-conjugated secondary antibody. All interphase cells showed a reactivity with juxtanuclear structures characteristic of the Golgi apparatus (Figures 2A-2C). Staining of the same cell structures was also observed in murine Swiss 3T3, P19, and AtT-20 cell lines (not shown). By contrast, no immunoreactivity was seen in any of the cell types incubated with the preimmune serum (not shown). Interestingly, the peri-nuclear distribution of DLK/MUK/ZPK was disrupted in mitotic cells in a manner consistent with a Golgi localization. During metaphase, when cells are rounded, DLK/MUK/ZPK was localized to vesicle-like structures uniformly distributed throughout the cytoplasm, whereas in early telophase an intense signal was detected in the centrosomal region of the mitotic spindle (Figures 2A and 2D-2F).
To provide additional evidence for the localization of DLK/MUK/ZPK to the Golgi region, we carried out immunohistochemical studies with DLK/MUK/ZPK antiserum on NIH 3T3 cells that were exposed to brefeldin A (BFA). BFA is a lipophilic fungal toxin that induces segregation of Golgi stacks into the endoplasmic reticulum and collapses of the trans-Golgi network on the microtubule-organizing center (Pelham 1991). As shown in Figures 3A and 3B, treatment of NIH 3T3 cells with BFA for 60 min leads to a nearly complete dispersion of DLK/MUK/ZPK staining into the cytoplasm. This disruptive effect of BFA on DLK/MUK/ZPK localization was entirely reversible. After removal of the drug from the culture medium, DLK/MUK/ZPK recovered its normal Golgi distribution in the perinuclear region of the cells (Figure 3C). Treatment of NIH 3T3 cells with ethanol, the solvent of BFA, had no effect on the perinuclear distribution of DLK/MUK/ZPK (Figure 3D). Location of DLK/MUK/ZPK in NIH 3T3 cells was also examined at the ultrastructural level by immunoelectron microscopy. Because initial attempts to localize DLK/MUK/ZPK by immunogold labeling did not give satisfactory results, we used as an alternative approach a pre-embedding immunoperoxidase technique for electron microscopic localization of DLK/MUK/ ZPK (Figure 4). As expected from the light microscopic immunocytochemistry, specific labeling for DLK/MUK/ZPK was found in the area of the Golgi complex (Figures 4A and 4C). Apparently, this labeling was associated with membranes of the stacked Golgi cisternae and associated vesicles. No staining was observed in the lumen of Golgi vesicles (Figure 4D). Negative control with preimmune serum gave no signal (Figure 4B).
Co-purification of DLK/MUK/ZPK with the Golgi Apparatus in Fractionated Cell Extracts
To confirm at the biochemical level the predicted Golgi association of DLK/MUK/ZPK, NIH 3T3 cells were fractionated using a self-forming Percoll gradient protocol that allows good resolution of plasma membranes, Golgi apparatus, endoplasmic reticulum, and lysosomes (Morand and Kent 1986). Each fraction was then assayed by immunoblotting with DLK/MUK/ZPK antiserum and with antisera to α-mannosidase II (Man II) and G-protein β-subunits, two marker proteins representative of the Golgi apparatus and plasma membrane, respectively. As shown in Figure 5, Western blot analysis revealed that DLK/MUK/ZPK and the Golgi marker Man II had overlapping fractionation profiles. Both proteins were particularly enriched in the heaviest fractions of the Percoll gradient and a considerable proportion of them co-purified in fractions 9 and 10. A mutually exclusive enrichment of DLK/MUK/ZPK and Man II was seen in fractions 8 and 11 respectively, but the significance of this observation is not known. The plasma membrane, as demonstrated by G-protein (β-subunit distribution, was predominantly found in fractions 2-4, in which no DLK/MUK/ZPK was detected (Figure 5). These results, together with the localization studies, strongly support the association of DLK/MUK/ZPK with the Golgi complex in NIH 3T3 cells.
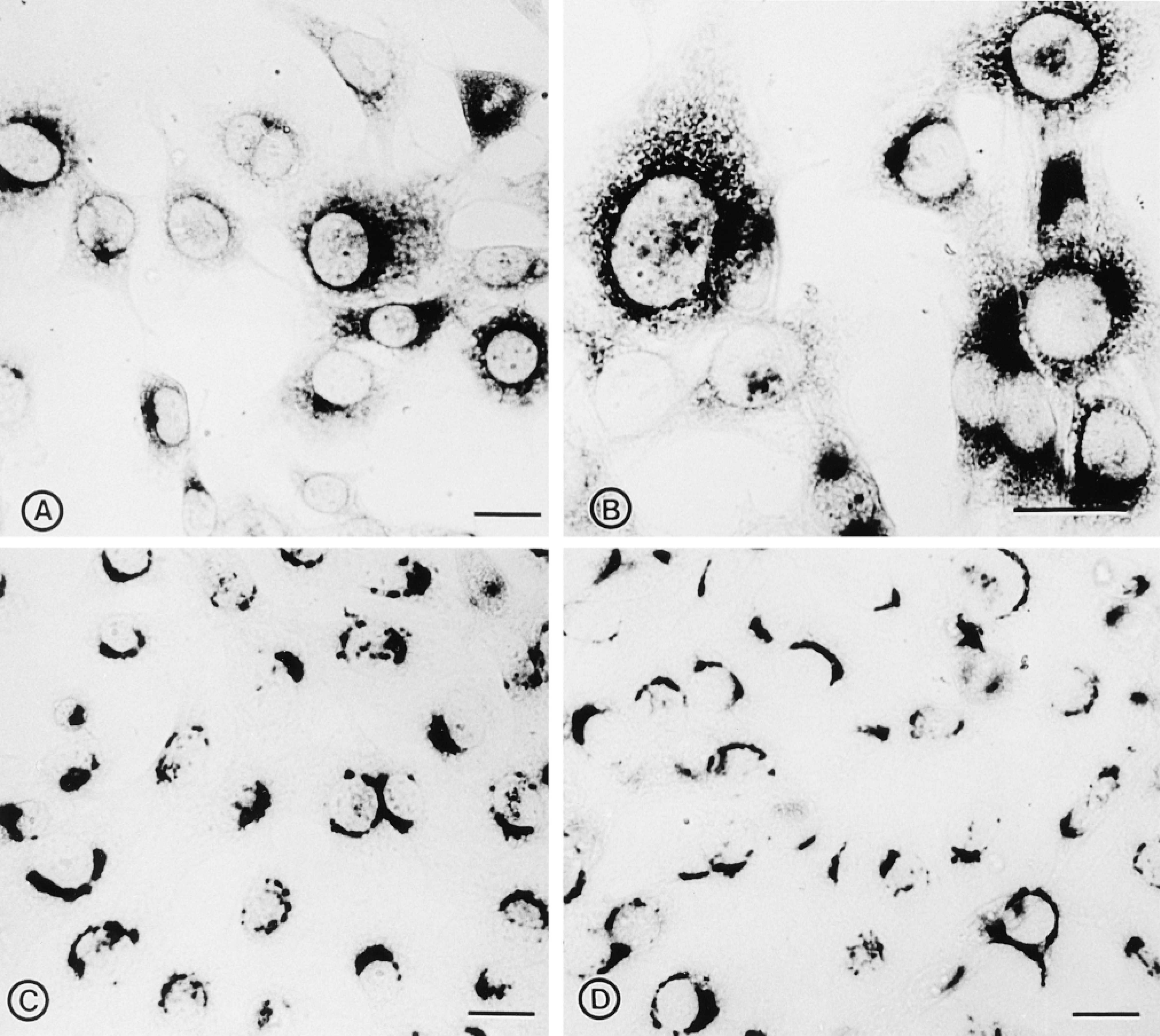
Effect of BFA on the distribution of DLK/MUK/ZPK. (A,B) NIH 3T3 cells grown on coverslips were treated with 2 μg/ml BFA for 60 min to break down the Golgi complex, fixed in methanol/acetone, and stained with DLK/MUK/ZPK antiserum. Treatment with BFA disrupted the juxtanuclear staining of DLK/MUK/ZPK. (C) NIH 3T3 cells were incubated for 60 min in the presence of 2 μg/ml BFA. At the end of the incubation period, cells were washed extensively and placed in drug-free culture medium for 60 min before fixation and staining with DLK/MUK/ZPK antiserum. The disruptive effects of BFA on the localization of DLK/MUK/ZPK are reversible after removal of the drug. (D) Control NIH 3T3 cells were grown in culture medium in the presence of BFA solvent only (ethanol) and immunolabeled with DLK/MUK/ZPK antiserum. Bars = 20 μm.
Membrane Association and Topology of DLK/MUK/ZPK
The nature of the association of DLK/MUK/ZPK with Golgi membranes was investigated using different extraction procedures, followed by Western blot analysis. As shown in Figure 6A, all DLK/MUK/ZPK immu noreactivity was associated with the membrane fraction prepared from NIH 3T3 cells (Figure 6, Lanes 1 and 2). This immunoreactivity could not be removed from particulate fractions even after treatment with 1 M NaCl, 1 M KCl, or 0.1 M Na2CO3, pH 11.5 (Figure 6, Lanes 3-8). When total cell membranes were subjected to Triton X-114 extraction, which allows discrimination between membrane-associated and integral membrane proteins, DLK/MUK/ZPK entirely partitioned into the aqueous phase (Figure 6, Lanes 9 and 10). These resuits suits therefore indicate that DLK/MUK/ZPK is a peripheral protein strongly associated with Golgi membranes.
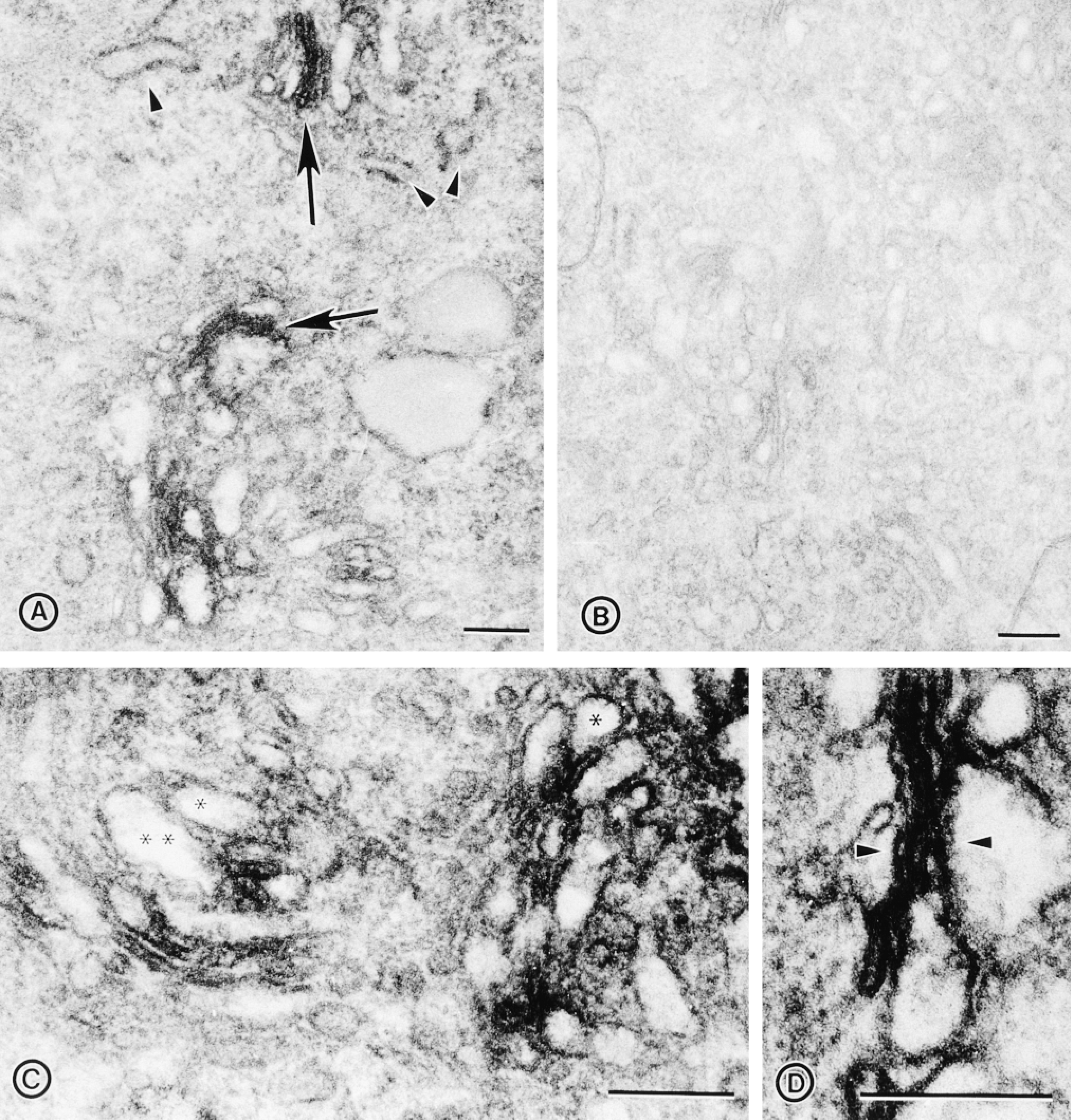
Electron microscopic immunoperoxidase localization of DLK/MUK/ZPK in NIH 3T3 cells. (A) Immunoperoxidase localization of DLK/ MUK/ZPK in NIH 3T3 cells, showing distribution of the reaction product on membranes of the stacked Golgi cisternae (arrows). No peroxidase reaction is seen in the endoplasmic reticulum (arrowheads). (B) Control cells incubated with preimmune serum. (C) High-magnification view of cells stained with DLK/MUK/ZPK antiserum shows absence of labeling in the lumen of vesicles located in the Golgi area (asterisks). (D) High-magnification view of cells stained with DLK/MUK/ZPK antiserum, showing association of the immunoperoxidase reaction product with Golgi stack membranes (arrowheads). Bars = 0.25 μm.
Next, to determine the topology of DLK/MUK/ZPK in Golgi membranes, particulate fractions prepared from NIH 3T3 cells were subjected to trypsin digestion in the presence or absence of a detergent. Figure 6B shows that DLK/MUK/ZPK was completely digested by trypsin even in the absence of a detergent (upper panel). Under the same conditions, Man II, an integral membrane protein facing the Golgi lumen, was resistant to digestion (Figure 6B, lower panel). Man II immunoreactivity was lost only when digestion was carried out in the presence of a detergent (Figure 6B, lower panel). Taken together, these results suggest that DLK/MUK/ZPK is a peripheral protein anchored to the cytoplasmic face of the Golgi complex.
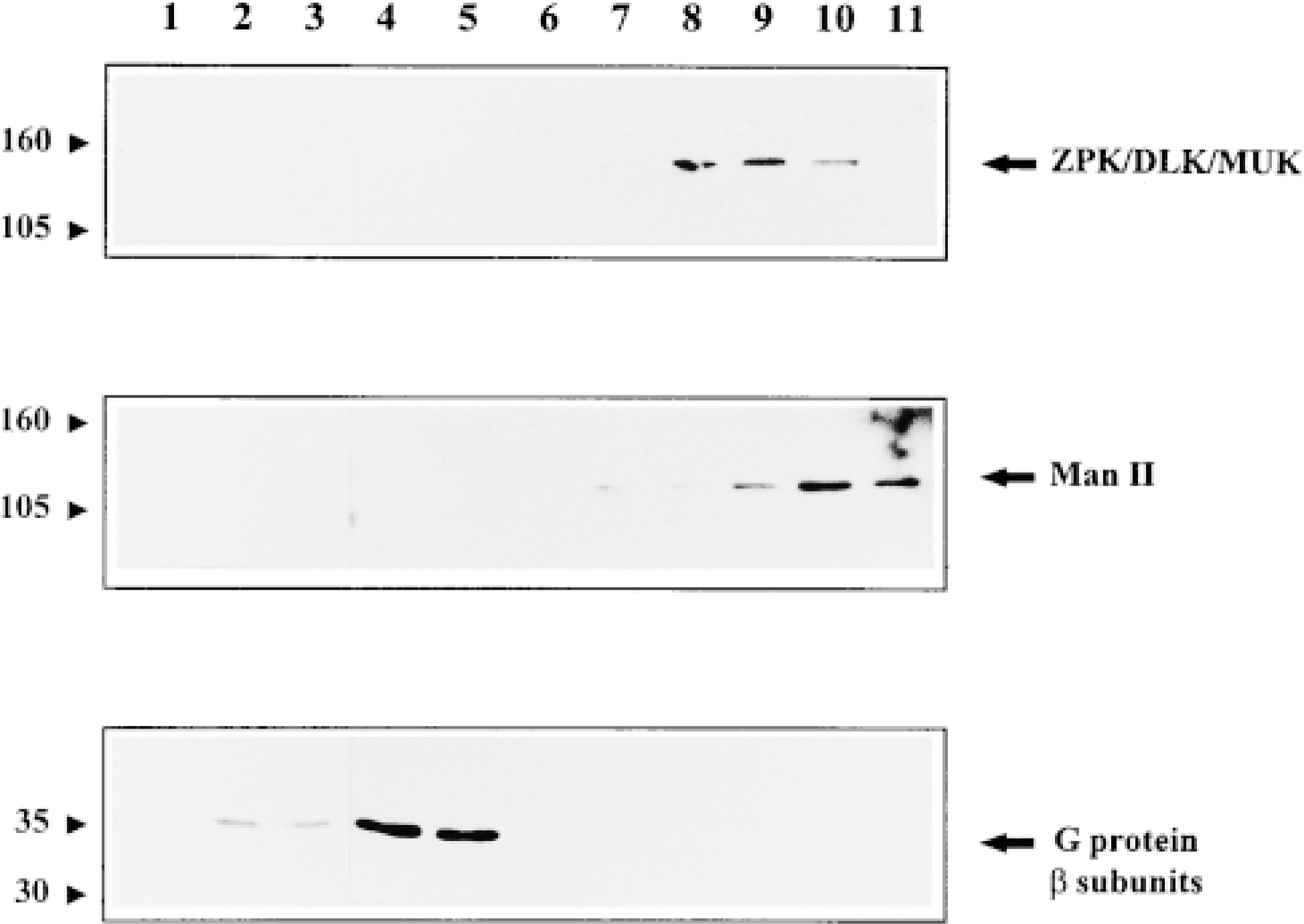
Subcellular distribution of DLK/MUK/ZPK. An NIH 3T3 cell homogenate was fractionated by centrifugation on self-forming Percoll gradient as described in Materials and Methods. Equal amounts of proteins (200 μg) from each fraction were subjected to SDS-PAGE, followed by immunoblot analyses with antiserum to either DLK/MUK/ZPK, Man II, or G-protein β-subunits.
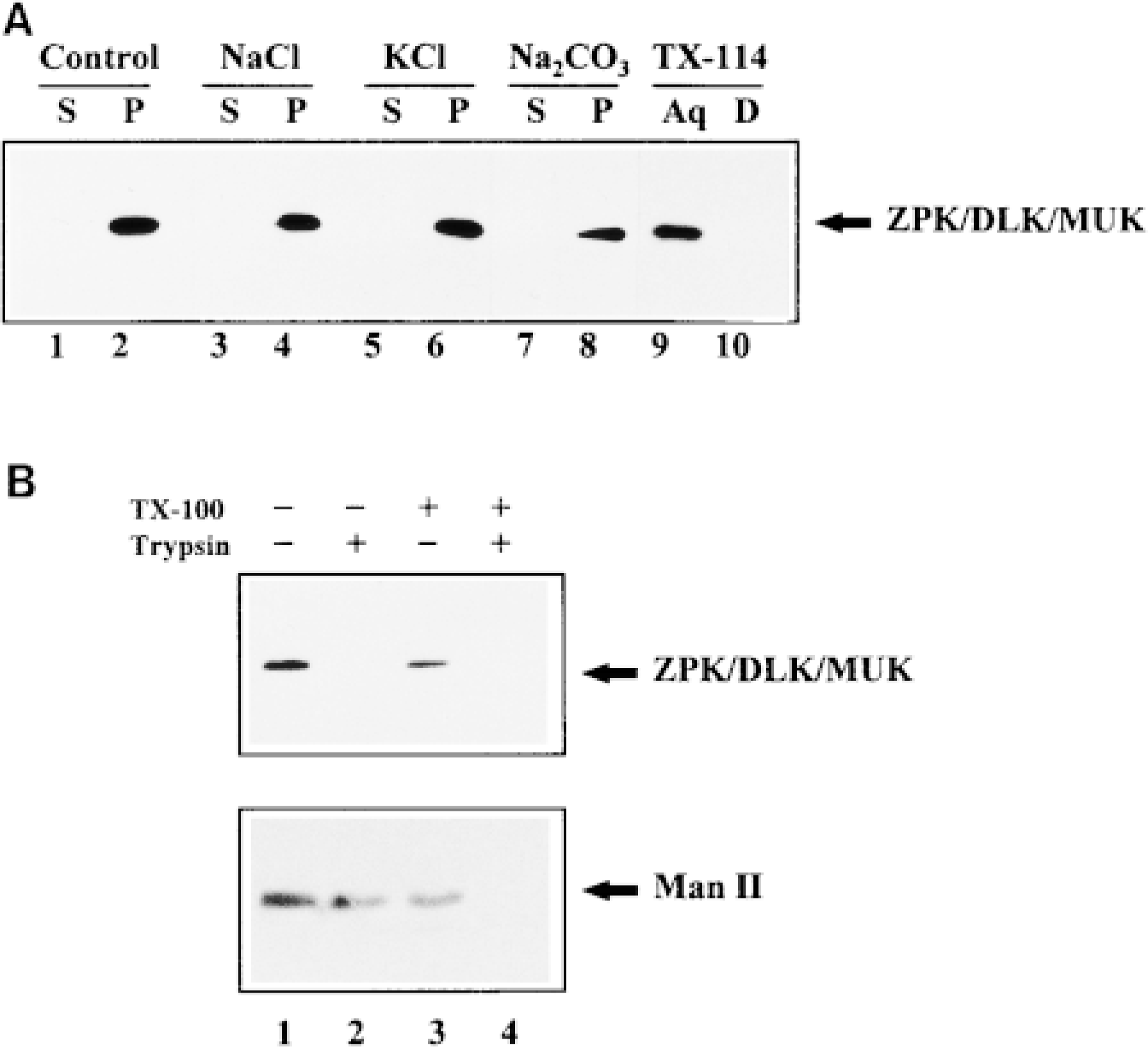
Association and topology of DLK/MUK/ZPK with Golgi membranes. (A) Particulate fractions (100,000 × g pellet) from NIH 3T3 cells were treated with control buffer (Lanes 1 and 2), 1 M NaCl (Lanes 3 and 4), 1 M KCl (Lanes 5 and 6), and 0.1 M Na2CO3, pH 11.5 (Lanes 7 and 8), for 30 min at 4C and then separated by centrifugation (100,000 × g for 1 hr) into supernatants (S) and pellets (P). Membrane pellets were resuspended in a volume of PBS buffer containing 1% Triton X-100 equal to that of supernatants and further extracted at 4C for 30 min. 50 μl of the supernatant and membrane fractions was then subjected to SDS-PAGE, followed by immunoblotting with DLK/MUK/ZPK antiserum. NIH 3T3 cell membrane fractions were also treated with Triton X-114 for 10 min on ice and partitioned into an aqueous (Aq, Lane 9) and a detergent (D, Lane 10) phase. Equal volumes of these fractions were processed as described above. (B) Particulate fractions from NIH 3T3 cells were incubated without (Lanes 1 and 3) or with (Lanes 2 and 4) 0.1 μg/ml trypsin in the absence (Lanes 1 and 2) or presence (Lanes 3 and 4) of 1% Triton X-100 for 20 min at 4C. Each sample was then adjusted to equal amounts of protease inhibitors and detergent and immunoprecipitated with antisera to DLK/MUK/ZPK or Man II. Immunocomplexes were fractionated on SDS-PAGE and Western blotted using antisera either to DLK/MUK/ZPK (upper panel) or Man II (lower panel).
Discussion
An antiserum against DLK/MUK/ZPK was produced to examine, for the first time, its endogenous subcellular distribution in cultured cells. The specificity of this antiserum was demonstrated on cell extracts prepared from transfected COS-1 cells and from wild-type NIH 3T3 and Swiss 3T3 fibroblasts. On the basis of these observations, which provide strong evidence for the suitability of this antiserum, the localization of DLK/MUK/ZPK was then investigated in NIH 3T3 cells by a combination of microscopic and biochemical experimental approaches. The results of these studies demonstrate that DLK/MUK/ZPK is specifically localized to the Golgi apparatus. We further found that DLK/MUK/ZPK has two of the properties expected for a peripheral and cytoplasmically disposed Golgi membrane protein. It partitioned solely into the aqueous phase of Triton X-114 extraction and was completely digested with trypsin under conditions in which the integrity of Man II was retained. In view of these findings, we propose that DLK/MUK/ZPK is a peripheral protein bound to the cytoplasmic face of Golgi membranes in NIH 3T3 cells. Support for this idea is provided by the observation that the amino acid sequence of DLK/MUK/ZPK has neither a putative signal peptide nor a hydrophobic membrane-spanning domain (Blouin et al. 1996). Recently, DLK/MUK/ZPK has also been reported to exist in both a cytosolic and a plasma membrane-bound form in rat brain synaptosomes (Mata et al. 1996). Although the significance of this discrepancy is not known at present, it may reflect differences in the cell context and/or the microenvironment.
Because DLK/MUK/ZPK behaves like a hydrophilic protein, as suggested by Triton X-114 extraction, its association with Golgi membranes may be mediated via binding to the cytoplasmic tail of an integral Golgi membrane protein. Like other members of the MLK family, DLK/MUK/ZPK contains domains that are beleived to be involved in protein-protein interactions, including several putative proline-rich SH3 binding motifs and a leucine/isoleucine zipper domain. These motifs may contribute to the interaction or association of DLK/MUK/ZPK with a specific integral membrane protein. Recently, it was demonstrated that MLK2/MST and MLK3/SPRK/PTK1 interacts in vitro with the small GTP binding proteins Rac and Cdc42, which are known to activate the JNK/SAPK and p38 MAPK pathways (Teramoto et al. 1996; Nagata et al. 1998). Although the functional significance of these interactions remains to be determined, recent studies have shown that small GTP binding proteins may function not only as regulators of signal transduction pathways but also as targeting molecules for protein kinases (Peter et al. 1996).
The finding that DLK/MUK/ZPK is localized to the Golgi complex in NIH 3T3 fibroblasts raises the intriguing possibility that this protein kinase plays a regulatory role in vesicle biogenesis and/or transport. Intracellular trafficking through the eukaryotic secretory pathway is a complex process controlled by the action of multiple mechanisms, including phosphorylation events (Davidson et al. 1992). Although the identity of the protein kinases involved in this regulation is still largely unknown, there is growing evidence suggesting an essential role for protein kinase A (PKA) and protein kinase C (PKC) in various vesicular transport processes, such as constitutive (Westermann et al. 1996; Muniz et al. 1997) and regulated exocytosis (Buccione et al. 1994; Robin et al. 1998) and transcytosis (Cardone et al. 1994; Hansen and Casanova 1994). Exactly how these kinases regulate vesicular traffic is unresolved, but recent studies have shown that several PKC family members are associated with Golgi membranes (Goodnight et al. 1995; Lehel et al. 1995; Prestle et al. 1996), and two of them have been found to modulate constitutive transport processes on overexpression in NIH 3T3 cells (Lehel et al. 1995; Prestle et al. 1996).
Indirect support for the idea that other MLK family members may somehow contribute to Golgi function is provided by the findings of Nagata et al. (1998), who recently reported that MLK2/MST localizes to vesicle-like structures along microtubules in fibroblastic cells. Interestingly, using the two-hybrid cloning system, the same investigators identified several potential MLK2/MST interacting proteins, among which members of the KIF3 subfamily of kinesin superfamily of proteins were found. Kinesins constitute a subclass of microtubule-associated motor proteins that play critical roles in intracellular transport (Brady 1995; Lippincott-Schwartz 1998). They are believed to drive the movement of membrane-bounded organelles and Golgi-derived vesicles along microtubules from the center of the cell to the periphery. The observation that MLK2/MST interacts in vitro with members of the kinesin family complements our own results. Taken together, these findings are consistent with the possibility that the MLKs fulfill regulatory functions in vesicular transport processes.
In summary, we have demonstrated that endogenous DLK/MUK/ZPK is specifically localized to the Golgi apparatus in NIH 3T3 cells, in which it behaves like a peripheral membrane protein facing the cytosol. These observations raise the possibility that this protein kinase is involved in the processes of intracellular lipid and protein transport, although there is no experimental evidence at present. Studies addressing the identification of the protein partners and physiological targets of DLK/MUK/ZPK should help us to determine whether this kinase could contribute to secretion.
Footnotes
Acknowledgements
Supported by a grant from the Natural Sciences and Engineering Research Council of Canada. M.D. is the recipient of a studentship from the Natural Sciences and Engineering Research Council of Canada.
We thank Dr Adrien R. Beaudoin and Ms Sheila Mac-Lean for critical reading of the manuscript.