
Abstract
The past decade has witnessed unprecedented progress in elucidation of the complex problems of the biogenesis of peroxisomes and related human disorders, with further deepening of our understanding of the metabolic role of this ubiquitous cell organelle. There have been many recent reviews on biochemical and molecular biological aspects of peroxisomes, with the morphology and cytochemistry receiving little attention. This review focuses on the state-of-the-art cytochemical techniques available for investigation of peroxisomes. After a brief introduction into the use of the 3,3′-diaminobenzidine method for localization of catalase, which is still most commonly used for identification of peroxisomes, the cerium technique for detection of peroxisomal oxidases is discussed. The influence of the buffer used in the incubation medium on the ultrastructural pattern obtained in rat liver peroxisomes in conjunction with the localization of urate oxidase in their crystalline cores is discussed, particularly since Tris-maleate buffer inhibits the enzyme activity. In immunocytochemistry, quantitation of immunogold labeling by automatic image analysis enables quantitative assessment of alterations of proteins in the matrix of peroxisomes. This provides a highly sensitive approach for analysis of peroxisomal responses to metabolic alterations or to xenobiotics. The recent evidence suggesting the involvement of ER in the biogenesis of “preperoxisomes” is mentioned and the potential role of preembedding immunocytochemistry for identification of ER-derived early peroxisomes is emphasized. The use of GFP expressed with a peroxisomal targeting signal for the investigation of peroxisomes in living cells is briefly discussed. Finally, the application of in situ hybridization for detection of peroxisomal mRNAs is reviewed, with emphasis on a recent protocol using perfusion-fixation, paraffin embedding, and digoxigenin-labeled cRNA probes, which provides a highly sensitive method for detection of both high- and low-abundance mRNAs encoding peroxisomal proteins.
Keywords
C
Because the DAB technique for localization of catalase, a marker enzyme of peroxisomes, is still the most commonly used cytochemical method for their investigation and one that is often used in an initial morphological survey for detection and identification of this organelle, it is treated first.
The DAB Technique for Localization of Peroxisomal Catalase
The peroxidase substrate 3,3′-diaminobenzidine was originally introduced by Graham and Karnovsky (1966) for the localization of horseradish peroxidase (HRP) used as a tracer. In the course of a joint project with M. J. Karnovsky on the endocytic uptake of HRP in liver (Fahimi and Karnovsky 1968), we noted that DAB simultaneously stained several organelles with endogenous peroxidase activity, i.e., peroxisomes, due to peroxidatic activity of catalase (Fahimi 1968,1969), mitochondria, due to cytochrome oxidase activity (Seligman et al. 1968; Angermüller and Fahimi 1981), and the endoplasmic reticulum in Kupffer cells (Fahimi 1970), due to an endogenous peroxidase involved in prostanoid synthesis (Deimann et al. 1984). Although in the initial reports the exact mechanism of staining of peroxisomes with DAB was controversial (Hirai 1968; Novikoff and Goldfischer 1969), the inhibition of the staining reaction by 3-amino-1,2,4-triazole indicated the involvement of peroxisomal catalase (Fahimi 1968, 1969). This notion was confirmed by the subsequent use of crystalline beef liver catalase as a tracer and its intravascular detection by the same method used for staining of peroxisomes (Venkatachalam and Fahimi 1969). The optimal conditions for staining of catalase with DAB were analyzed in vitro with crystalline beef liver catalase and with isolated rat liver peroxisomes (Herzog and Fahimi 1974,1976; LeHir et al. 1979). These studies revealed that “fixation” of catalase with an aldehyde increases the peroxidatic activity of that enzyme and facilitates its detection by DAB (Herzog and Fahimi 1974). This principal of tissue “fixation” with an aldehyde can also be used for the distinction between catalase (which requires fixation) and peroxidases, which are usually highly sensitive to aldehyde fixation (Fahimi et al. 1976; Herzog and Fahimi 1976). The optimal conditions for detection of peroxisomal catalase with DAB are tissue fixation with an aldehyde, incubation with 1-2% DAB at high-alkaline pH of 10.5, at 37C or 45C, and a high concentration (0.15%) of hydrogen peroxide (LeHir et al. 1979; Angermüller and Fahimi 1981). These conditions have been used in a large variety of vertebrate and invertebrate systems and have consistently yielded excellent results (Figures 1a and 1b). In this context, the “universal” buffer of Teorell and Stenhagen, which has a broad range (pH 2-12) and a strong buffering capacity, has proved very useful (Fahimi 1975), although glycine-NaOH buffer can be used as well. This buffer and very similar incubation conditions to those mentioned above have also been applied for detection of peroxisomes in biopsies of patients suspected of peroxisomal disorders (Roels et al. 1987) or in Pex5-/- transgenic mice (Baes et al. 1997) with defects in biogenesis of peroxisomes (Figures 3a and 3b). For further details concerning the selective staining of peroxisomes with DAB and cytochemical distinction between different enzymes with peroxidase activity, see Deimann et al. (1991).
DAB-stained semithin sections have been used for morphometric assessment of the peroxisomal compartment and its alterations in response to xenobiotics (Fahimi et al. 1982; Beier and Fahimi 1986, 1987, 1991; Beier et al. 1988; Lindauer et al. 1994). For this purpose, semithin sections stained for catalase are analyzed by light microscopic automatic image analysis and an excellent correlation is found between the time-consuming electron microscopic morphometry and the highly efficient automatic image analysis (Beier and Fahimi 1987,1992).
Techniques for morphological investigation of peroxisomes
Cerium Technique for Detection of Oxidase Activity in Peroxisomes
The cerium technique is presently the method of choice for light and electron microscopic detection of peroxisomal oxidases, although alternative methods based on reduction of ferricyanide and tetrazolium salts were used in earlier years (for a review of the early literature on peroxisomal oxidases see Angermüller 1989; Robinson et al. 1991). Cerium was introduced by Briggs et al. (1975) for detection of H2O2 generated by NADH-oxidase in granulocytes, and a modification of their method was adopted for the localization of peroxisomal oxidases in yeast (Veenhuis et al. 1976). They reported, however, that in rat liver, because of the extreme sensitivity of peroxisomal oxidases to fixation, the best results were obtained in unfixed tissue (Veenhuis and Wendelaar Bonga 1979). Somewhat better results were reported by others in kidney and central nervous system (Arnold et al. 1979; Hand 1979; Arnold and Holtzman 1980). Almost a decade later, Angermüller and Fahimi (1986) reported the appropriate processing conditions for fixation and visualization of the oxidase activities in different vertebrate tissues, consisting of a short perfusion-fixation with 0.25% glutaraldehyde in PIPES buffer, pH 7.4, followed by incubation in 3 mM cerium chloride with specific substrates (e.g., urate, glycolate, or
The final reaction product of cerium chloride with H2O2 is cerium perhydroxide, which is electron-dense (Briggs et al. 1975) but lacks sufficient contrast in transmitted light. A procedure for light microscopic visualization of the cerium reaction product using DAB-nickel or -cobalt chloride was introduced by Angermüller and Fahimi (1988b). This method and slight modifications of it introduced subsequently (Gossrau et al. 1989) have been applied widely for the localization of various (also non-peroxisomal) oxidases (for review see van den Munckhof 1996). Because the reaction products of cerium and DAB can both be detected in the reflectance mode of the confocal laser scanning microscope (Robinson and Batten 1989,1990), there is some potential in the application of this technique for the future investigation of peroxisomes and their oxidases (Halbhuber et al. 1998).
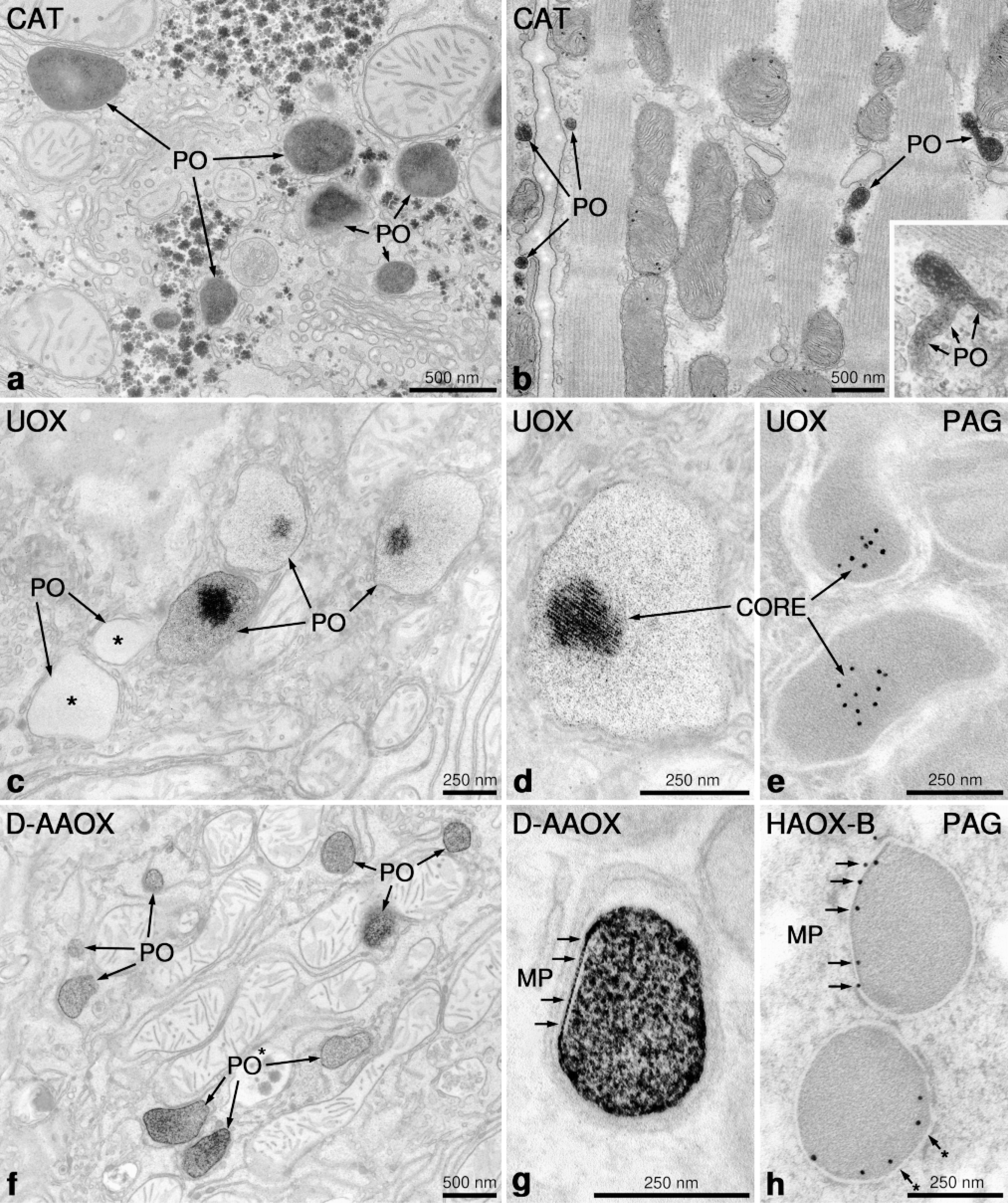
Application of cytochemical techniques for localization of peroxisomal enzyme activity. (a) Rat liver peroxisomes (PO) stained with alkaline DAB technique (pH 10.5) for detection of peroxidatic activity of catalase (CAT). (b) Peroxisomes (PO) in rat myocardium stained for the localization of catalase (CAT). Note the elongated tubular form of some peroxisomes, with evidence of branching (inset) reminiscent of the peroxisomal reticulum found in regenerating rat liver (Yamamoto and Fahimi 1987). (c,d) Peroxisomes in rat liver stained for the localization of urate oxidase (UOX) activity by the cerium technique. Note distinct staining of the crystalline core only in some peroxisomes. Other peroxisomes (∗) completely lack the cerium reaction product, clearly demonstrating the heterogeneity of peroxisomes (PO) within the same hepatocyte. (e) Immunocytochemical localization of urate oxidase by the protein A-gold technique (PAG). Note the exclusive localization of gold particles to the core region. (f) Rat hepatic peroxisomes (PO) stained for D-amino acid oxidase activity using D-proline (D-AAOX) as substrate. Note the heterogeneity in the intensity of staining between adjacent peroxisomes (*) within the same cell. (g, h) Peroxisomes with marginal plates (MP). In g the MP in rat kidney stained for D-amino acid oxidase (DAA-OX) appears as negative contrast below the peroxisomal membrane (arrows). In h, the immunolabeling with an antibody to α-hydroxy acid oxidase-B reveals the curved lentiform appearance of marginal plates in a feline kidney (arrows) (Courtesy Dr. K. Zaar).
Cytochemistry with cerium, together with immunoelectron microscopy, has revealed that some oxidases form distinct subcompartments in the matrix of peroxisomes. Therefore, in addition to the electron-dense polytubular cores of UOX,
Immunocytochemical Techniques
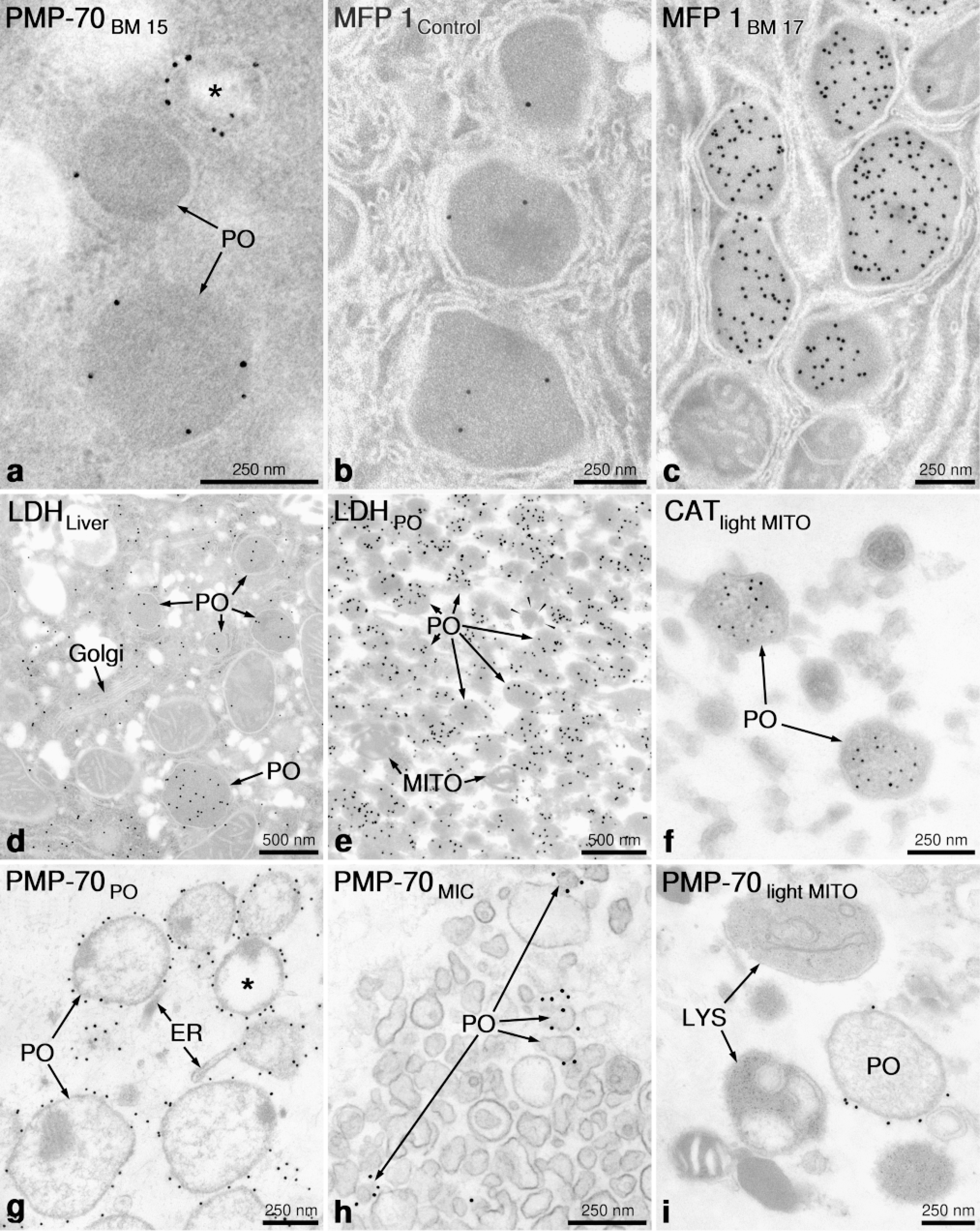
During the past two decades, immunolabeling techniques have gained a great deal in importance, becoming the principal tools for morphological investigation of peroxisomes. With the availability of antibodies against most peroxisomal proteins and development of suitable protocols for processing of cells and tissues, immunofluoresence and immunoelectron microscopy are now almost routine procedures in peroxisome research. Moreover, those techniques have become indispensible tools in diagnosis and research of peroxisomal diseases revealing, e.g., the presence of the so-called peroxisomal ghosts in the liver of patients with generalized peroxisome disorders (Espeel et al. 1995b). The term “ghost” was proposed by Santos et al. (1988), who found by fluorescence microscopy immunoreactive peroxisomal membrane proteins in cell lines from patients with Zellweger syndrome, lacking detectable peroxisomes. By immunoelectron microscopy, the ghosts consist of small (100-200-nm) vesicles and double-membraned loops, which are immunoreactive with antibodies to peroxisomal membrane proteins but in most instances lack matrix proteins (Espeel et al. 1995b). Similar membranous structures were reported earlier in the liver of rats treated with a new hypocholesterolemic drug (BM 15766), which induces peroxisomal membrane proteins (PMP) and organelle proliferation without concomitant elevation of the β-oxidation enzymes (Baumgart et al. 1989,1990). In pericentral hepatocytes of rats treated with this drug, double membrane loops and ring- and hook-shaped structures in close vicinity or direct continuity with peroxisomes are observed, which can be labeled with the antibody to the 70-kD PMP in the absence of immunoreactivity for catalase or any other matrix proteins (Figure 2a). Therefore, these membranes represent proliferated peroxisomal structures without a luminal content, because of the lack of simultaneous increase in the synthesis of matrix proteins. A similar situation exists in the majority of peroxisomal biogenesis disorders in which the transport of some or all matrix proteins is defective, although some membrane proteins, such as the PMP-70, are present at normal levels (Lazarow and Moser 1995). Similar membranous structures were also found recently in the livers of PEX5-/- mice, the first mouse model of the Zellweger syndrome, generated by the inactivation of PEX5, the gene of the receptor for the import of most peroxisomal matrix proteins containing the peroxisomal targeting signal 1 (Baes et al. 1997). Those mice lack morphologically identifiable peroxisomes and exhibit the typical biochemical abnormalities of patients with Zellweger syndrome (Lazarow and Moser 1995). The mice exhibit, in addition, evidence of intrauterine growth retardation, are severely hypotonic at birth, and die within 72 hr after birth. Figure 3 illustrates the localization of catalase by the DAB technique and immunoelectron microscopy in the liver of PEX5-/- (Figures 3b and 3d) and control mice (Figures 3a and 3c). Whereas peroxisomes are clearly visualized by both methods in control livers, they are absent in PEX5 -/mice. Moreover, in the latter animals catalase is expressed and detectable in the cytoplasm of liver cells, with some heterogeneity in the level of expression in neighboring hepatocytes (Cells A, B and C in Figure 3b and Cells A and B in Figure 3d). This mosaic pattern in catalase expression is somewhat reminiscent of a mosaic pattern with respect to occurrence of peroxisomes in some hepatocytes described in patients with Zellweger syndrome (Espeel et al. 1995a). The exact mechanism for the development of this mosaicism remains unknown, but it appears likely that the PEX5 -/-mouse model could be useful for elucidation of this problem and for investigation of the pathogenesis of severe organ anomalies associated with disorders of peroxisome biogenesis.
Application of immunoelectron microscopy for detection of peroxisomal protein antigens. (
Whereas in the first immunocytochemical studies ferritin- and peroxidase-labeled antibodies were used (Yokota and Nagata 1974; Yokota and Fahimi 1981, 1982), at present the immunogold technique is the method of choice (Fahimi et al. 1996a; Usuda et al. 1996). The protocols developed by Litwin et al. for the immunolabeling of peroxisomes in rodents (Litwin et al. 1984), and humans (Litwin et al. 1987) and modifications thereof are widely used (Espeel et al. 1995b). The preservation of the antigenicity is excellent in ultrathin frozen sections, and most enzymes and proteins involved in peroxisomal sterol metabolism (Krisans 1996) have been detected by this method (Keller et al. 1985,1986,1989; Krisans et al. 1994). Nevertheless, many peroxisomal proteins are sufficiently robust, withstanding the resin-embedding conditions, that LR White, Lowicryl K4M, and Unicryl are frequently used in different laboratories (Espeel at al. 1995b; Fahimi et al. 1996b; Yokota and Okada 1997).
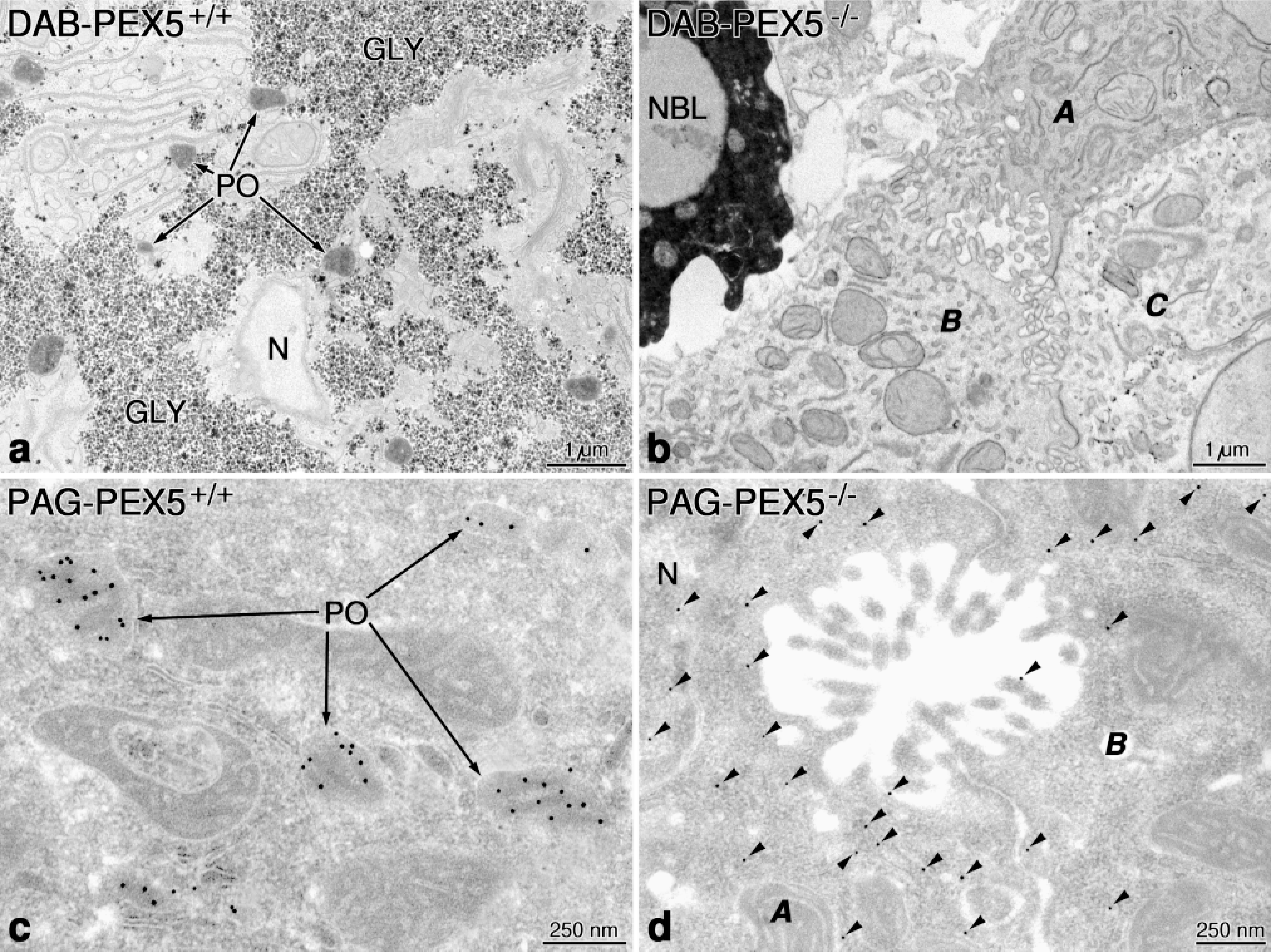
Sections from the livers of PEX 5-/- and control mice processed for localization of catalase by the DAB technique and by immunocytochemistry for catalase. Note the prominent staining of peroxisomes (PO) in control mice (a,c) and their absence in Zellweger mice (b,d). In PEX 5-/- mouse, catalase labeled by gold particles (arrowheads) is found in the cytoplasm and also in the nucleus (N) of hepatocytes (d). Note the heterogeneity in the intensity of catalase staining and immunolabeling in different hepatocytes (Cells A, B, and C in b and Cells A and B in d). NBL, normoblast.
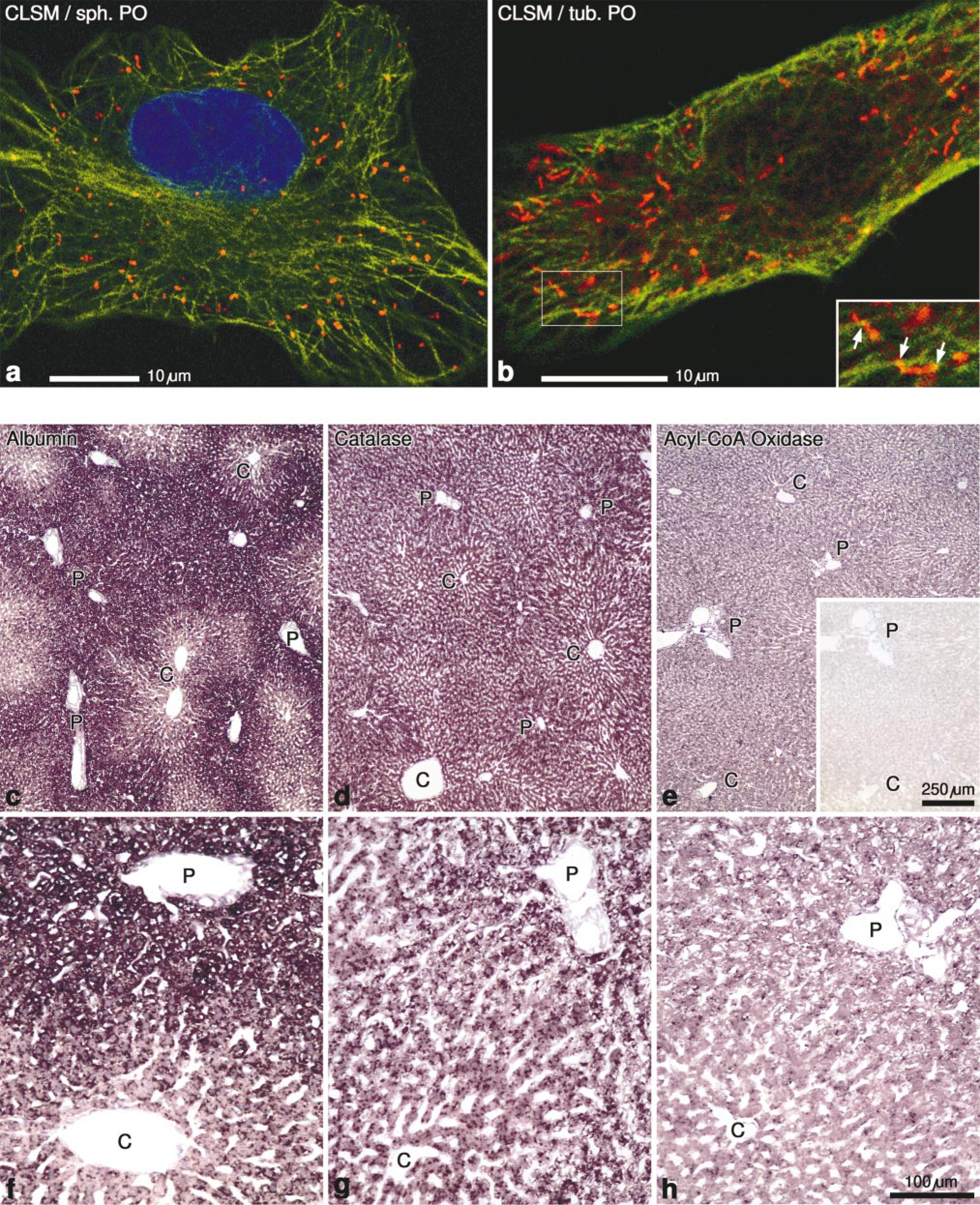
(a,b) Confocal laser scanning micrographs (CLSM) of HepG2 cells labeled with antibodies to catalase (rhodamine), and tubulin (fluorescein), overlaid with a 2-photon micrograph showing nuclei counterstained with DAPI. Note the spherical form of peroxisomes in a in contrast to their tubular form in b. The contact sites between microtubules and peroxisomes are rather rare (arrows in inset to b). (c-h) In situ hybridization of rat liver sections with digoxigenin-labeled cRNA probes. (c,f) Albumin mRNA distribution with stronger periportal (P) staining. (d,g) The mRNA for catalase is uniformly distributed in the rat liver lobule. (e,h) The mRNA for acyl-CoA oxidase is slightly stronger in periportal hepatocytes (P) than in the pericentral region (C) of the liver lobule. (e,inset) Sense control with no evidence of staining.
Immunogold labeling in combination with automatic image analysis permits quantitative assessment of alterations of proteins in peroxisomes in response to treatment of rodents and cell cultures with different agents (Beier and Fahimi 1992). Thus, peroxisome proliferators (Reddy and Lalwani 1983) increase significantly the immunogold labeling density (number of gold particles per μm2 of peroxisome matrix) for β-oxidation enzymes but not for UOX or D-amino acid oxidase (Beier et al. 1988). The increase in immunolabeling density is dose-dependent and is more pronounced in pericentral hepatocytes of the liver lobule (Lindauer et al. 1994), where the corresponding mRNAs encoding the β-oxidation enzymes are also initially elevated (Schad et al. 1996a,b; Baumgart 1997; Baumgart et al. 1997). The induction of β-oxidation enzymes is species-dependent and is observed mainly in rodents, but not in primates and humans (Fahimi et al. 1993; Reddy and Mannaerts 1994). This is apparently related to the structure and level of the peroxisome proliferator-activated receptor-α (PPAR-α) which mediates the xenobiotic-induced response (Tugwood et al. 1998).
Recently, an isoform of lactate dehydrogenate (LDH) was detected in rat hepatic peroxisomes by cell fractionation and immunocytochemistry (Baumgart et al. 1996). Although LDH is primarily a cytosolic protein, by the immunogold technique, it can be clearly localized to the peroxisome matrix (Figure 2d), and this is also confirmed in isolated purified peroxisomes (Figure 2e), with catalase serving as control for identification of peroxisomes (Figure 2f). It was suggested that peroxisomal LDH is involved in shuttling reducing equivalents (NADH) produced by fatty acid β-oxidation out of peroxisomes into the cytoplasm. This notion is supported by the marked increase of peroxisomal LDH after the induction of peroxisomal β-oxidation by bezafibrate, a peroxisome proliferator (Baumgart et al. 1996).
The application of immunoelectron microscopy to isolated subcellular fractions has led to the disovery of peroxisomes with lower buoyant density than the regular particles. Whereas the regular peroxisomes in rat liver band at a density of 1.23-1.24 g/cm2, in regenerating rat liver particles with lower density (1.20 g/cm2 and lower) were found which in pulse-labeling studies exhibited a higher initial rate of incorporation of radioactivity, suggesting that they could be precursor particles to regular peroxisomes (Lüers et al. 1993). Similar conclusions were obtained in other experimental models (Heinemann and Just 1992; Wilcke et al. 1995) suggesting a “maturation” of peroxisomes. Recent observations suggest that in differentiating HepG2 cells in culture, peroxisomes indeed undergo a maturation process, becoming larger and acquiring new enzymes (e.g., branched chain acyl-CoA oxidase) and that PPAR-α is probably involved in this process (Stier et al. 1998). The development and application of a reliable method for preembedding immunolabeling of peroxisomes in isolated subcellular fractions (Baumgart 1994; Völkl et al. 1996), have been quite important in the recent progress in biochemical characterization of this organelle. The high resolving power and precision of this technique using an antibody to PMP-70 is shown in peroxisomal (Figure 2g), microsomal (Figure 2h), and light mitochondrial fractions (Figure 2i). It would have been impossible to identify the small particles in the microsomal fraction as derivatives of peroxisomes without the immunolabeling (Figure 2h).
Although the concept of derivation of peroxisomes from the endoplasmic reticulum, originally advanced by Alex Novikoff (for a review see Novikoff and Novikoff 1982), was refuted by the mid-1980s (Lazarow and Fujiki 1985), recently evidence has been accumulating that the endoplasmic reticulum may be involved in the early stages of de novo formation of peroxisomes. Indeed, some peroxins, the gene products involved in biogenesis of peroxisomes (e.g., Pex15p and Pex16p) may possibly be inserted first into ER membranes before budding off and fissioning to form peroxisomes (for reviews see Erdmann et al. 1997; Faber et al. 1998; Kunau and Erdmann 1998; Titorenko and Rachubinski 1998). For exact identification of such “preperoxisomes,” which are involved in the transport of those peroxins from the ER to peroxisomes, the above-mentioned immunocytochemical preembed ding techniques appear to be highly promising. Moreover, the recently developed methodology of immunofreeflow and immunomagnetic isolation of peroxisomes (Völkl et al. 1997,1998; Lüers et al. 1998) may be suitable to isolate those ER-derived “vesicles” once specific antibodies against the cytosolic domains of those early peroxins are available.
The application of immunofluorescence has revealed the existence of tubular peroxisomes measuring up to several microns, next to small spherical particles in HepG2 cells (Figures 4a and 4b) (Schrader et al. 1994,1996). Similar elongated peroxisomes are also observed in myocardium (Hicks and Fahimi 1977) (Figure 1b) and small intestine (Roels et al. 1991). The tubular peroxisomes in HepG2 cells are induced by specific growth factors (e.g., nerve growth factor, NGF) and by polyunsaturated fatty acids, and disappear by fission, giving rise to small spherical particles (Schrader et al. 1998). For proper preservation of tubular peroxisomes, it is important to fix the cells with paraformaldehyde because fixation with ethanol or methanol/acetone can fragment the elongated peroxisomes, giving rise exclusively to small spherical particles in immunofluorescence preparations (Schrader et al. 1995).
In contrast to tubular mitochondria (Johnson et al. 1980) and lysosomes (Swanson et al. 1987; Kobayashi and Robinson 1991; Robinson and Luo 1992), which collapse into spherical forms after depolymerization of the microtubule system, the tubular peroxisomes maintain their shape and are even increased in frequency after treatment with nocodazole or colcemid (Schrader et al. 1996). Prolonged exposure of cells (9-12 hr) to those drugs, however, induces the formation of focal aggregates of peroxisomes, demonstrating the important role of microtubules for the uniform distribution of peroxisomes throughout the cytoplasm. The association of microtubules and peroxisomes is difficult to visualize in routine double-immunofluorescence preparations, but by confocal laser scanning microscopy focal contacts can be clearly observed (Figure 4b and inset). Such contacts have also been described by electron microsopy (Rapp et al. 1996; Baumgart 1997). In vivo studies using either transfection with green fluorescent protein (GFP) expressed with the peroxisome targeting signal SKL (Wiemer et al. 1997) or microinjection of FITC-luciferase (Rapp et al. 1996) have revealed that 90-95% of peroxisomes are almost stationary, whereas the remaining 5-10% exhibit an ATP-dependent saltatory movement with a speed of 0.05-1 μm/sec over a distance of 10-12 μm. Observations in mitotic cells have revealed that peroxisomes are randomly distributed and are not associated with spindle microtubules, suggesting that peroxisomal inheritance is stochastic rather than ordered (Wiemer et al. 1997).
As mentioned, expression of the GFP with the peroxisomal targeting signal 1 (SKL) has been used successfully for in vivo labeling of peroxisomes in yeast and mammalian cells (Wiemer et al. 1997). By coupling the fluorescence of GFP to photo-oxidation of DAB, Monosov et al. (1996) could identify the intracellular location of GFP also by electron microscopy. This would provide a reliable tool for combined light and electron microscopic analysis of targeting of proteins to peroxisomes.
In Situ Hybridization (ISH)
The main application of ISH in peroxisome research has been for chromosomal localization of genes encoding peroxisomal proteins and for assessment of the cellular and tissue-specific distribution of their corresponding mRNAs. According to the published reports on chromosomal assignment studies in mammals the “peroxisomal genes” are not clustered on specific chromosomes but rather are found on different ones.
There have been only a few studies on application of ISH for the assessment of tissue-specific and cellular distribution of “peroxisomal mRNAs” (for a recent review see Baumgart et al. 1997). This could be due to the relatively low levels of expression of most mRNAs encoding peroxisomal proteins, thus requiring highly sensitive ISH techniques. Indeed, all initial reports on detection of mRNAs for catalase (Reimer and Singh 1990), 3-keto-acyl-CoA thiolase (Bout et al. 1990), multifunctional protein I (Rao et al. 1994), D-amino acid oxidase (Koibuchi et al. 1995), and PMP-70 (Pollard et al. 1995) were carried out with 35 S-labeled cRNA probes, which have a relatively high sensitivity but a low level of cytological resolution. A sensitive and elegant assay for expression of the acyl-CoA oxidase gene in different cells and tissues in response to treatment with peroxiome proliferators was introduced recently by Reddy and associates, utilizing the human acyl-CoA oxidase gene promoter to direct the expression of E. coli β-galactosidase (lacZ) in transgenic mice (Pan et al 1996; Reddy and Chu 1996). After treatment, tissues were fixed and stained for β-galactosidase, revealing heavy staining not only in well-known responsive tissues (liver, kidney, and intestine) but also in reproductive organs, Langerhans islands in pancreas, macrophages of spleen and lung, and Purkinje cells and neurons in the brain. Recently, Schad et al. (1996a) introduced a novel, highly sensitive protocol for ISH using digoxigenin-labeled probes and paraffin sections for detection of mRNAs encoding peroxisomal proteins. Thus, mRNAs coding for catalase and UOX, representing high-abundance transcripts, as well as mRNAs for multifunctional protein I (Schad et al. 1996a), acyl-CoA oxidase (Fahimi et al. 1996b), and PMP-70 (Schad et al. 1996b), representing low-abundance transcripts, have been localized in rat liver and kidney. In all instances the specific mRNAs are confined to cells containing large numbers of peroxisomes: the liver parenchymal cells (Figures 4d, 4e, 4g, and 4h) and proximal cortical tubules, particularly the P3 segments in kidney (Schad et al. 1996a,b; Baumgart et al. 1997). The cells in portal tracts, including the bile duct epithelial cells and the sinusoidal cells in liver (Figures 4g and 4h), and the distal tubules, collecting ducts, and glomeruli in kidney are negative (not shown). Figures 4d and 4e illustrate the difference in abundance of mRNAs for catalase and palmitoyl-CoA oxidase in normal rat liver and in comparison with a parallel control section hybridized with a sense probe, confirming the specificity of the reaction (Figure 4e, inset). In comparison with albumin mRNA, which is clearly expressed at a higher level in periportal regions of the normal liver lobule (Figures 4c and 4f), the catalase transcripts are uniformly distributed (Figures 4d and 4g), while palmitoyl-CoA oxidase is slightly more abundant in periportal hepatocytes (Figures 4e and 4h). After treatment with peroxisome proliferators, the mRNA levels for β-oxidation enzymes markedly increase in pericentral hepatocytes (Schad et al. 1996a,b; Fahimi et al. 1996c; Baumgart et al. 1997), whereas the transcripts for UOX become reduced. The selective increase of transcripts for (β-oxidation enzymes corresponds to the similar elevations in levels of proteins for those enzymes (Lindauer et al. 1994) and could be related to the higher levels of PPAR-alpha in the same region of the liver lobule (Huang et al. 1995; Fahimi et al. 1996c).
The main features of the protocol used by Schad et al. (1996a,b) are (a) perfusion-fixation with 4% depolymerized paraformaldehyde and 0.05% glutaraldehyde, (b) paraffin embedding and routine preparation of 2-4 μm sections, (c) careful control of the deproteination step with proteinase K, and (d) use of digoxigenin-labeled cRNA probes and signal detection with alkaline phosphatase-labeled Fab fragments and NBT-BCIP staining. The latter step can be prolonged up to 24 hr at 37C, increasing the sensitivity of the method to the level of the radioactive techniques. Nevertheless, the control sections incubated with sense probes remain completely negative (Figure 4e, inset) confirming the specificity of this method. With the recent development of amplification protocols to increase the sensitivity of the nonradioactive ISH techniques (Bobrow et al. 1989; Speel et al. 1998), it may soon become possible to analyze the alterations of peroxisomal mRNAs under physiological regulatory conditions, such as hunger and feeding, and in pathological conditions, such as those induced by inflammatory mediators, e.g., by tumor necrosis factor-α (Beier et al. 1997).
Footnotes
Acknowledgements
Supported by SFB-352 (project C7), SFB 601 (project B1), and grants FA 146/3-1, Ba1155/1-3, all from the Deutsche Forschungsgemeinschaft, Bonn-Bad Godesberg, and by a grant from the European Community DGXII-PL96-3569.
We thank all our colleagues and collaborators at the Institute for Anatomy and Cell Biology at the University of Heidelberg for their many helpful suggestions and discussions. In particular, Drs Alfred Völkl, Sabine Angermüller, Kurt Zaar, and Konstantin Beier have contributed over the years to many of the important results from our laboratory summarized here. We also thank Gabrielle Burger (Leica; Heidelberg, Germany) for assistance with preparation of CLSM and two photon microscopic images (Figures 4a and 4b) and Dr Kurt Zaar for the micrograph on immunocytochemical localization of α-hydroxy acid oxidase B (marginal plates; ![]() ). The technical assistance of Gabi Krämer and Heike Steininger is gratefully acknowledged.
). The technical assistance of Gabi Krämer and Heike Steininger is gratefully acknowledged.