
Abstract
Difficulties in specific detection of transfected DNA in cells represent an important limitation in the study of the gene transfer process. We studied the cellular entry and fate of a plasmid DNA complexed with a cationic lipid, Vectamidine (3-tetradecyl-amino-N-tert-butyl-N-tetradecylpropionamidine) in BHK21 cells. To facilitate its detection inside the cells, bromodeoxyuridine (BrdU) was incorporated into plasmid DNA under conditions that minimize plasmid alteration. BrdU was localized in cells incubated with Vectamidine/BrdU-labeled plasmid DNA complexes by immunogold labeling and electron microscopy (EM). Labeling was predominantly associated with aggregated liposome structures at the surface of and inside the cells. EM observations of cells transfected with Vectamidine/DNA complexes showed that the liposome/DNA aggregates accumulate in large vesicles in the cell cytosol. On the other hand, using rhodamine-labeled Vectamidine and revealing BrdU with FITC-conjugated antibodies permitted simultaneous detection in the cells of both components of the complexes with confocal laser scanning microscopy. The DNA and lipids co-localized at the surface of and inside the cells, indicating that the complex is internalized as a whole. Our results show that the BrdU-labeled plasmid DNA detection system can be a useful tool to visualize exogenous DNA entry into cells by a combination of electron and confocal microscopy.
Keywords
C
The goal of this work was to follow the cellular entry of plasmid DNA complexed to a new cationic lipid (3-tetradecylamino-N-tert-butyl-N′-tetradecylpropion-amidine: Vectamidine). Vectamidine was used because of its higher transfection efficiency and lower toxicity to the cells (Ruysschaert et al. 1994; El Ouahabi et al. 1996, 1997). BrdU was incorporated into plasmid DNA and was visualized by confocal or electron (EM) microscopy. Both confocal and EM consistently support endocytosis as the main pathway of cationic liposome/DNA complex entry into cells.
Materials and Methods
Reagents
Vectamidine (previously described as diC14-amidine) was purchased from Biotech Tools (Brussels, Belgium). V-(lissamine rhodamine B sulfonyl) phosphatidylethanolamine (Rh-PE) was from Avanti Polar Lipids (Alabaster, AL). Bromodeox-yuridine triphosphate (BrdU), bovine serum albumin (BSA), and other phospholipids were from Sigma Chemical (St Louis, MO). Anti-bromodeoxyuridine (anti-BrdU) monoclonal antibody and DNA polymerase I were from Boehringer (Mannheim, Germany). FITC-conjugated anti-mouse polyclonal antibodies and the DNase I/DNA polymerase mixture (cat no. N5000) were from Amersham (Poole, UK).
Cell Culture
BHK21 cells obtained from the ATCC (Rockville, MD) were grown in Glasgow MEM supplemented with 5% FBS, 10% tryptose phosphate broth, 2 mM glutamine, and antibiotics. Cells were kept at 37C in a humidified 5% CO2 atmosphere and were passaged every 2 days.
Labeling of Plasmid DNA with BrdU
The plasmid DNA pCMV-CAT [containing the chloramphenicol acetyl transferase (CAT) bacterial gene under the control of the CMV immediate early promoter/enhancer] was kindly provided by Dr. R. Debs and was previously described (Zhu et al. 1993). The pCMV-CAT plasmid DNA was extracted by the alkaline lysis method (Sambrook et al. 1989) and was further purified using a Qiagen Kit according to the manufacturer's instructions. To label the pCMV-CAT plasmid DNA with BrdU, we used a modified nick-translation technique (El Ouahabi et al. 1997). Briefly, a 100-μl reaction mixture was prepared to contain 50 mM Tris-HCl, pH 7.5, 10 mM MgSO4, 0.1 mM DTT, 50 μg/ml BSA, 0.1 mM each nucleotide (except dTTP), 10 μg DNA, 2 μl (10 U/μl) DNA polymerase I, 2 μl of the DNA polymerase I/DNase I mixture, and 0.1 mM BrdU. Under these conditions, the low DNase I:DNA ratio and the increased DNA polymerase I:DNase I ratio allowed the incorporation of BrdU into the plasmid DNA without significant alteration of plasmid integrity, as assessed by agarose gel electrophoresis.
Liposome Preparation
Vectamidine liposomes were prepared by the ethanol injection method as described (El Ouahabi et al. 1996). Rhodamine-labeled Vectamidine liposomes were prepared as described (El Ouahabi et al. 1996), with some modifications. Briefly, 20 μl of ethanol containing 1 mg of Vectamidine and 1% (molar) Rh-PE was diluted into 1 ml of prewarmed HBS buffer under vigorous vortexing.
Vectamidine/DNA Complex Preparation
Plasmid DNA or BrdU-labeled plasmid DNA and Vectamidine liposomes were separately diluted in 25 μl of HBS buffer before being mixed and incubated for 10-15 min at room temperature (RT). Unless otherwise indicated, 1 μg DNA and 2 μg Vectamidine was used. This leads to spontaneous formation of a quantitative complex which was previously characterized (Ruysschaert et al. 1994; El Ouahabi et al. 1997). The complexes were further diluted in 1 ml of serum-free medium before being added to cells.
Immunofluorescence Staining and Confocal Microscopic Analysis
For immunofluorescence experiments BHK21 cells were plated at 2 × 104 per well in 8-well Lab-Tek chamber slides (Nunc; Roskilde, Denmark). BrdU-labeled DNA 0.2 μg and Rh-PE-labeled Vectamidine 0.4 μg were each diluted in 10 μl of HBS, mixed, and incubated at RT for 10-15 min. The complexes were further diluted with 80 μl of serum-free medium and incubated with cells for 5, 15, 30, 60, and 120 min. At the end of the incubation, the cells were washed three times with serum-free medium, once with PBS, and then fixed with freshly prepared 2% paraformaldehyde in PBS at RT for 30 min. Fixed cells were incubated in 2 N HCl for 1 hr at 37C and then the acid was neutralized by incubation in 0.1 M borate buffer, pH 8.5, twice for 5 min. Cells were washed with PBS three times for 5 min and incubated with the anti-BrdU antibody at 3 μg/ml in PBS containing 0.5% BSA for 1 hr at RT. After two 5-min washes with PBS and one 5-min wash with PBS containing 0.5% BSA, the cells were incubated with goat anti-mouse IgG coupled to FITC (diluted 1:100 in PBS containing 0.5% BSA) for 1 hr at RT. Cells were then washed three times for 5 min with PBS and once for 5 min with water. The chamber-forming structures were removed and the slide was mounted in Vectashield mounting medium (Vector Laboratories; Burlingame, CA). Several controls were carried out. When the acid treatment or the primary antibody was omitted, no fluorescence could be detected in the cell. Likewise, nontransfected cells or cells transfected with nonlabeled plasmid DNA complexed to liposomes were unlabeled. To further confirm the specificity of the anti-BrdU antibody detection, BrdU was incorporated into the nuclear DNA by metabolic labeling and the cells stained as described above. In this case, labeling was found exclusively within nuclei.
Cells were observed under a Zeiss Axiovert fluorescence microscope, and images were obtained using a laser scanning confocal microscope (MRC 1000; Bio-Rad, Hercules, CA) equipped with an argon-krypton laser and Comos software (Bio-Rad). Serial images of rhodamine and FITC fluorescence at 0.72-μm Z-intervals were recorded separately and then combined. Images were further analyzed using Imagespace software (Molecular Dynamics; Sunnyvale, CA).
Electron Microscopy
BHK21 cells were seeded onto 6-well plates at 500,000 cells/well and incubated overnight. Cells were treated with Vectamidine/DNA complexes for 2-3 hr, then washed and scraped off the plates. Cells were centrifuged at 350 × g for 3 min to form pellets. Small fragments of the various pellets were fixed for 30 min at RT in 2.5% glutaraldehyde in 0.1 M Sorensen's buffer (pH 7.4). After washing in buffer, cells were postfixed in 2% OsO4 for 30 min at RT, dehydrated in ethanol (30%, once for 10 min; 70%, three times for 10 min) and embedded in Epon 812 resin (70% ethanol:Epon, 1:1, twice for 10 min; 70% ethanol:Epon, 1:2, twice for 10 min; pure Epon, overnight). The polymerization was achieved at 60C. Ultrathin sections (90 nm) were placed on 200-mesh nickel grids and stained with uranyl acetate and Reynold's lead citrate. The samples were examined in a Jeol CX100 electron microscope at 60 KV.
Immunogold Staining
To follow the cellular entry and fate of the DNA, cells were seeded onto 24-well plates at 50,000 cells/well and incubated overnight. BrdU-labeled plasmid DNA was complexed with Vectamidine and incubated with cells for 5, 15, 30, 60, and 120 min. At the end of the incubation, cells were washed with PBS and fixed in situ for 120 min at 4C in 4% formaldehyde in 0.1 M Sorensen's buffer. After several washes in buffer, cells were dehydrated in graded ethanol solutions (30, 70, 95, 100%) and embedded in Epon. The resin was polymerized over 3 or 4 days at 40C. Detection of BrdU incorporated into plasmid DNA was performed as previously described (Thiry and Dombrowicz 1988). Briefly, ultrathin sections were prepared and mounted on gold grids, then incubated at RT in 5 N HCl for 30 min. The latter were incubated by floating them down on a drop of PBS containing goat normal serum diluted 1:30 and 1% BSA, then rinsed with PBS containing 1% BSA. The next step of the treatment was an incubation with monoclonal anti-BrdU antibody diluted 1:300 in PBS containing 0.2% BSA and normal goat serum diluted 1:50 for 4 hr at RT. After washing with PBS containing 1% BSA, the sections were incubated for 1 hr with goat anti-mouse IgG coupled to colloidal gold 10-nm diameter (Amersham) diluted 1:50 with PBS (pH 8.2) containing 0.2% BSA at RT. After washing with PBS containing 1% BSA, the sections were rinsed in bidistilled water and dried before being stained with uranyl acetate and lead citrate. Several controls were used. When nontransfected cells or cells transfected with liposomes alone or with BrdU-unlabeled DNA complexed with liposomes were used, the sections were devoid of label. Likewise, no label was observed when the primary antibody was omitted. Finally, gold lacking the antibody tag did not bind to the sections.
Results
Study of the Vectamidine/DNA Complex Entry into Cells by Electron Microscopy
BHK21 cells were incubated with Vectamidine/DNA complexes and ultrathin sections were prepared and processed for transmission electron microscopy (TEM) with uranyl acetate staining (for details see Materials and Methods). Vectamidine/DNA complexes were identified on the ultrathin sections by their high electron density and appeared as aggregations of ovoid structures (Figure 1, arrows) and filament networks (Figure 1, arrowheads). At 2-3 hr after incubation, these aggregates adhered to the cell surface or were taken up by the cells through an endocytic process (Figure 1A). In some instances, aggregates in the process of being engulfed by the cells were observed (an example is indicated by the large arrow in Figure 1A). Once in the cytosol, the Vectamidine/DNA aggregates appeared to be contained in large endocytic vesicles that were filled to some extent with the aggregates (Figure 1B).
Although Vectamidine/DNA complexes were identified by their high electron density, no specific labeling was associated with the DNA, and therefore the fate of the DNA after interaction of the complexes with cells could not be clearly established. To overcome this difficulty, BrdU-labeled DNA was complexed with Vectamidine and incubated with cells for various times (ranging from 5 to 120 min). Then the incorporated BrdU molecules were revealed by postembedding immunogold labeling. It should be emphasized that these experimental conditions are compatible with the in situ immunodetection of BrdU incorporated into the DNA but do not make possible a clear recognition of cell membranes. Labeling was associated with large aggregates consisting of variable electron-dense rings reminiscent of liposomes (Figures 2A-2D, arrows). More specifically, gold particles were preferentially located over filaments that extended from the rings (Figures 2A-2D, arrowheads), suggesting that they may be plasmid DNA molecules. The fact that these filaments were not observed when liposomes alone were incubated with cells supports this conclusion (not shown). At 15-30 min after incubation with cells, these Vectamidine/DNA aggregates were detected at the surface of a few cells (Figures 2A and 2B). For longer incubation times (2 hr), Vectamidine/DNA aggregates could be detected in the cytosol (Figures 2C and 2D). Aggregates that were in the process of being engulfed by the cells but were not fully internalized were also observed (an example is indicated by the large arrow in Figure 2C). We did not observe any gold label in the nucleus up to 24 hr after the initial incubation of cells with the complexes.
Altogether, these results (Figures 1 and 2) clearly demonstrate that the Vectamidine/DNA complexes are internalized by an endocytic process and that they contain the transfected DNA. However, we cannot exclude the possibility that fusion between the cationic liposome of the complex and the plasma membrane leads to entry of DNA directly into the cytoplasm. Membrane fusion is indeed considered to be a rapid process and therefore is difficult to monitor by EM (Zabner et al. 1995).
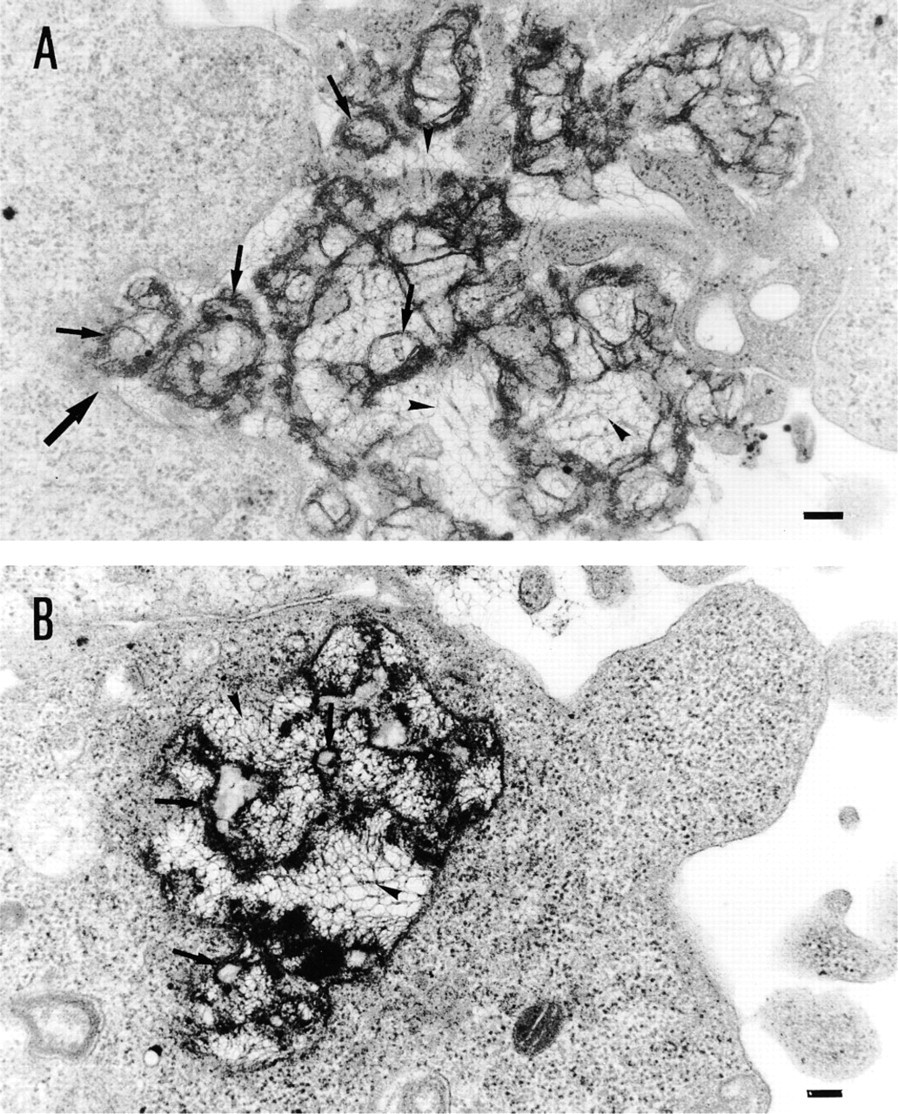
Visualization of the pathway of Vectamidine/DNA complex entry into cells by electron microscopy: transmission electron micrographs of BHK21 cells treated with Vectamidine/DNA complexes for 3 hr. Complexes were observed adhering to the cell surface (
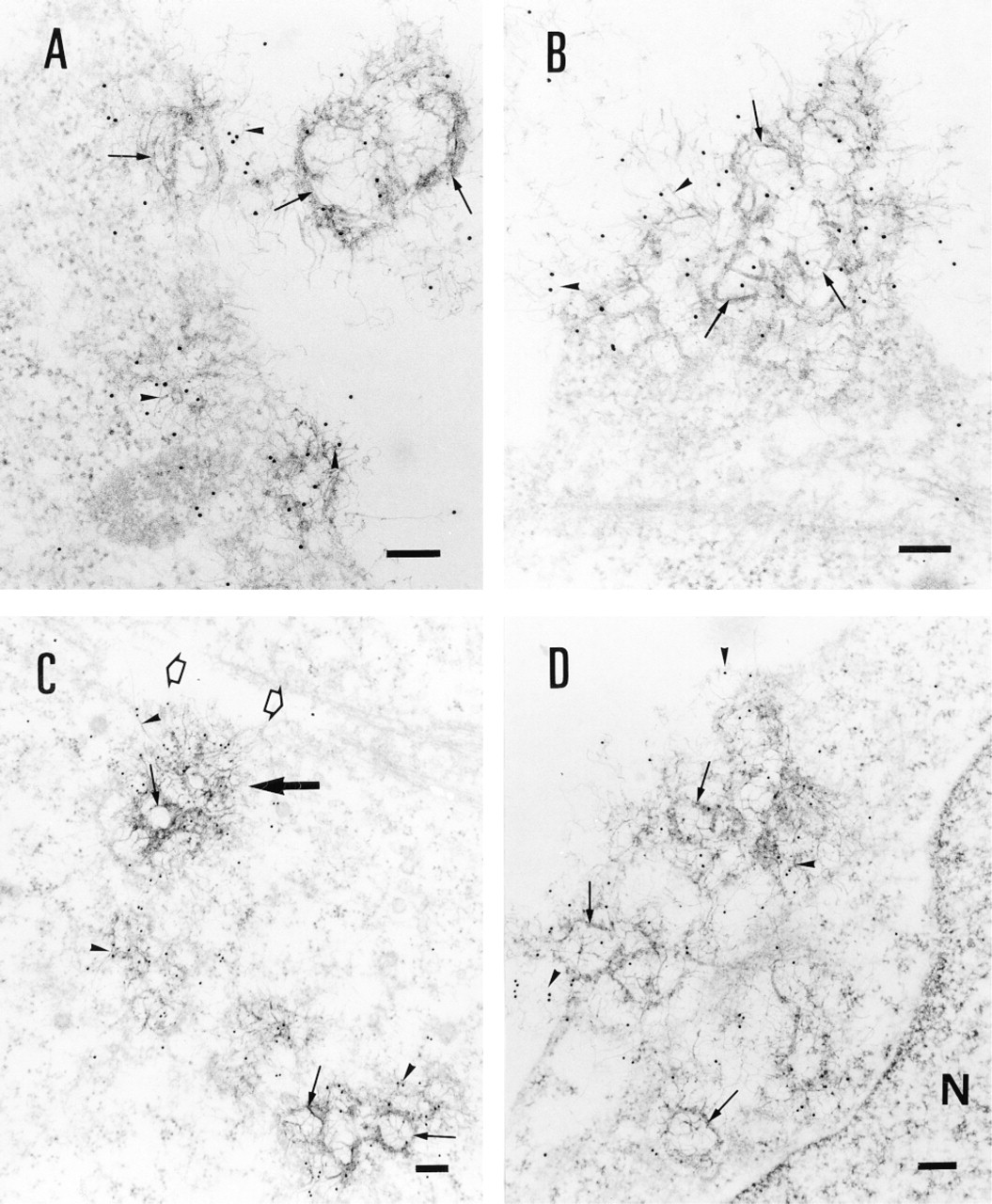
Immunogold detection of BrdU-labeled plasmid DNA in the cells. BHK21 cells were treated with Vectamidine/BrdU-labeled plasmid DNA complexes for different times and then ultrathin sections were prepared and BrdU revealed using immunogold labeling as described in Materials and Methods. Transmission electron micrographs showing complexes adsorbed at the cell surface (
Interaction of the Vectamidine/DNA Complexes with Cells: A Confocal Microscopic Study
To simultaneously visualize the cellular entry and the fate of Vectamidine and the DNA, BHK21 cells were incubated with complexes containing Rh-PE-labeled Vectamidine and BrdU-labeled plasmid DNA. After cell fixation, BrdU was revealed by immunofluorescence with an FITC-conjugated antibody. The fluorescence distribution inside cells was analyzed by confocal laser scanning microscopy. Liposome alone or DNA alone was red or green, respectively, whereas the DNA/liposome complex was yellow.
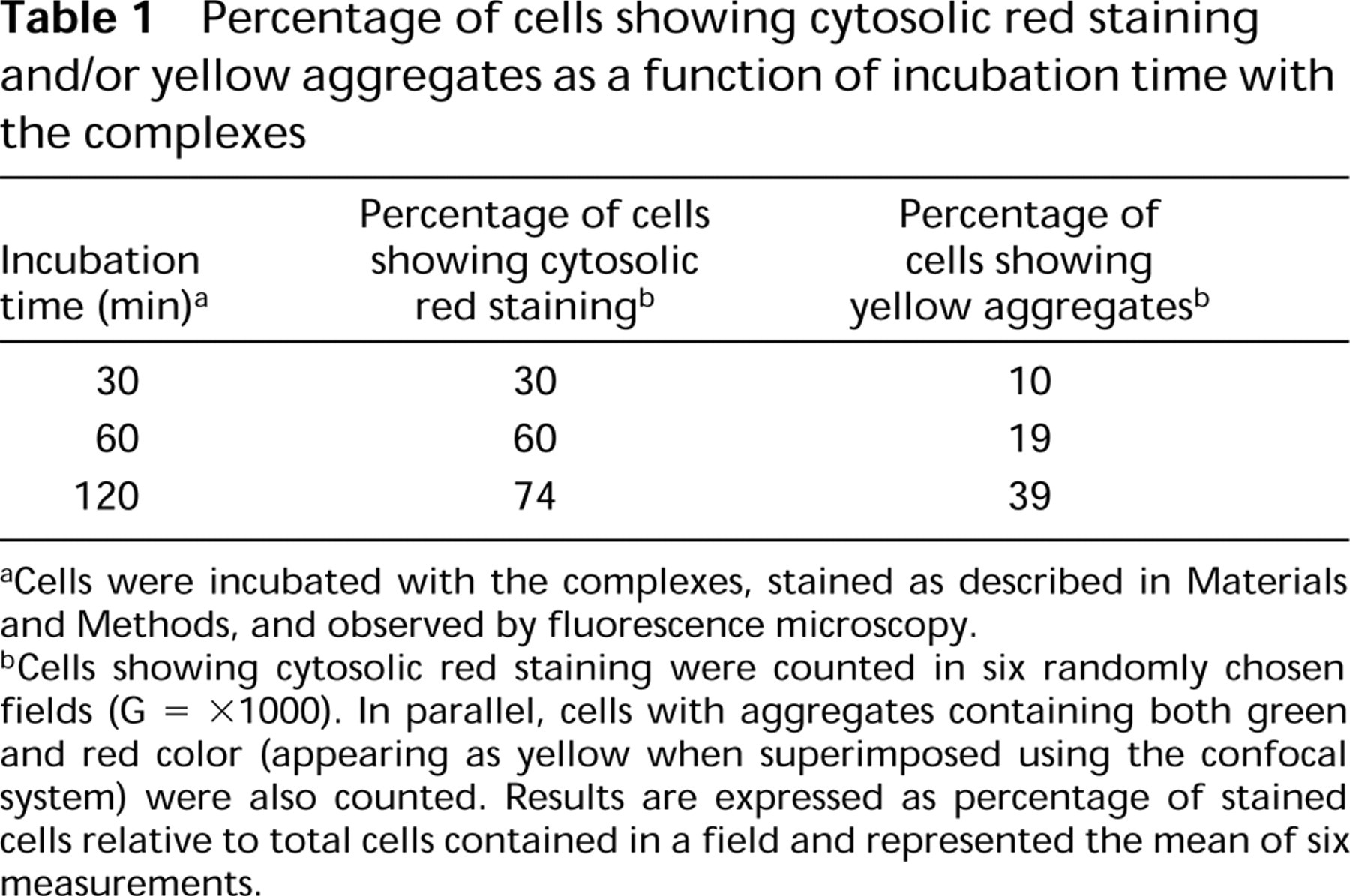
After short incubation times (15-30 min) of cells with the doubly labeled Vectamidine/DNA complex, yellow aggregates (corresponding to the co-localization of red-associated liposome and green-associated DNA colors) were detectable in intercellular spaces and at the surface of some cells. The fraction of yellow aggregate-associated cells increased with increasing incubation times to a maximum after 2 hr (Table 1). At this time, some yellow aggregates appeared inside the cells (Figure 3A, arrows), whereas others were adsorbed to the cell surface (Figure 3A, arrowhead). The cytosol became progressively red-stained with increasing incubation times (Figures 3A and 3B). After 2 hr of incubation with the complex, the majority of the cells showed a strong cytosolic red staining (Table 1). As seen in Figure 3B, red staining was more pronounced in the regions of cytosol where yellow aggregates were located, suggesting that the red staining was probably derived from the internalized complexes. The fact that the cell cytosol was red-stained suggests that, after entry of complexes into the cytoplasm, Vectamidine vesicles fuse with intracellular membranes, leading to redistribution through the intracellular membrane network. The DNA would be released in the cytoplasm and diluted so that its fluorescence becomes too weak to be detected (no visible green color). However, we failed to detect any significant red or green fluorescence inside the nucleus, even 12 hr after adding the complexes to cells (not shown).
The fact that intracellular aggregates were yellow supports the idea that the Vectamidine/DNA complex penetrates as a whole inside the cell before it dissociates. If fusion between Vectamidine liposomes and plasma membrane were the major event during complex-cell interaction, one would expect the plasma cell membrane to become red-labeled before the intracytoplasmic membrane network, which is not the case. No cell labeling was observed when Rh-PE-labeled neutral phosphatidylcholine liposomes were incubated with cells, confirming other work showing that Rh-PE is not a probe that can be transferred to cells by a trivial lipid exchange mechanism (not shown). When Bodipy-phosphocholine was used instead of Rh-PE to label Vectamidine liposomes, the same pattern of cell fluorescence distribution was observed (not shown), suggesting that the fluorescence distribution observed here reflected the behavior of the cationic lipid and not that of the fluorescent probe used.
Percentage of cells showing cytosolic red staining and/or yellow aggregates as a function of incubation time with the complexes
Cells were incubated with the complexes, stained as described in Materials and Methods, and observed by fluorescence microscopy.
Cells showing cytosolic red staining were counted in six randomly chosen fields (G = X1000). In parallel, cells with aggregates containing both green and red color (appearing as yellow when superimposed using the confocal system) were also counted. Results are expressed as percentage of stained cells relative to total cells contained in a field and represented the mean of six measurements.
Discussion
Cationic liposome-mediated gene transfer has become a widely used tool for in vitro and in vivo transfection of eukaryotic cells. This process involves the formation of a cationic liposome/DNA complex that efficiently interacts with the cell surface, leading to entry and expression of the exogenous DNA. However, events that occur between the initial interaction of the cationic liposome/DNA complexes with the cell surface and the expression of the transfected DNA are not yet understood. Difficulties in specific detection of the transfected DNA in cells represented an important limitation in the study of the gene transfer process. Here, we optimized a BrdU-based labeling technique that keeps the DNA as close as possible to its native form and makes it detectable by confocal and electron microscopy. Confocal and electron microscopy studies consistently converged towards an endocytosis mechanism as the main pathway for Vectamidine/DNA complex internalization by BHK21 cells. This conclusion was supported by several data. (a) Transmission electron microscopic observations of ultrathin sections from transfected cells revealed electron-dense complexes formed by aggregated liposomes and filament networks contained within endosome-like structures inside the cell. (b) Immunogold labeling of BrdU incorporated into plasmid DNA showed that the DNA is associated with the aggregated liposomes at the surface of and inside the cells. (c) Confocal microscopy showed that cationic liposome/DNA complexes enter the cell as a whole before dissociation. Zabner and colleagues (1995) and Friend and colleagues (1996) have also used EM and have imaged the DNA of complexes in the cell, but their approach is questionable because they incorporated gold beads into the complexes before incubation with the cell. Here, incorporation of the nucleotide analogue BrdU into plasmid DNA is performed under conditions that minimize plasmid alteration.
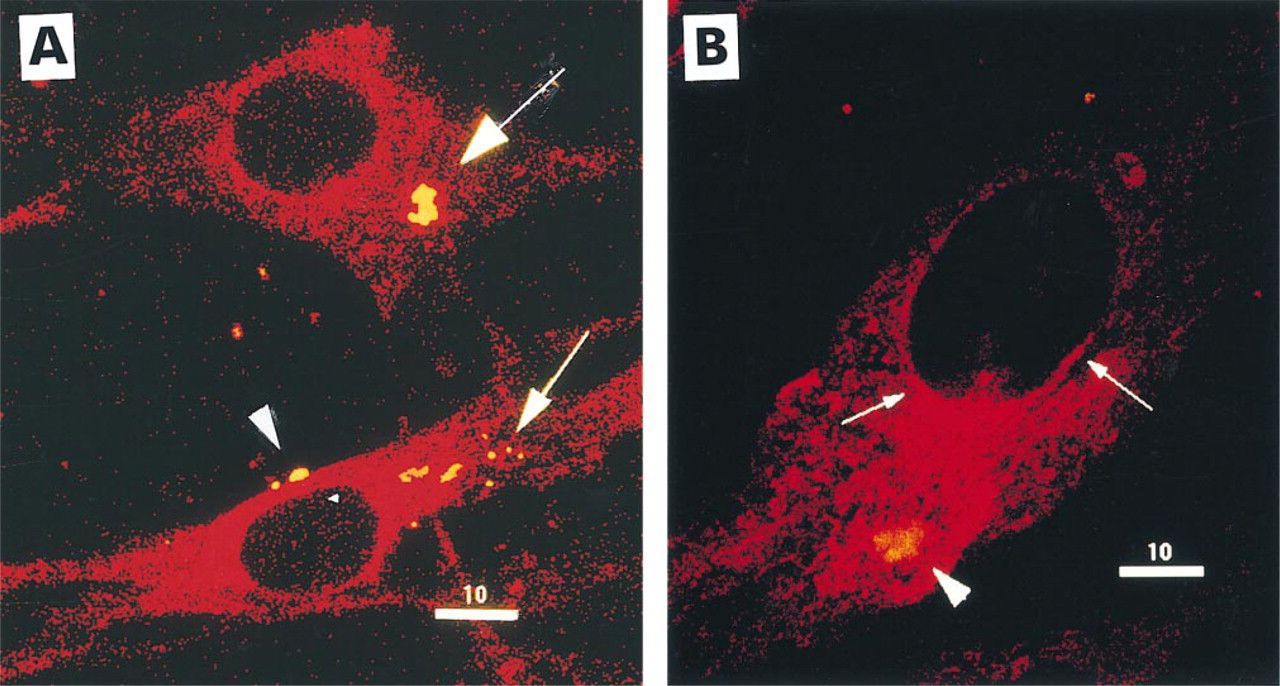
Simultaneous visualization of DNA and Vectamidine interaction with cells by confocal microscopy. Fluorescence confocal micrographs showing BHK21 cells treated for 2 hr with complexes of green-labeled BrdU/DNA (detected by anti-BrdU, FITC-stained) and redlabeled Vectamidine liposomes (containing rhodamine/PE). Yellow aggregates result from the superimposition of green (DNA) and red (liposomes). (
EM data showed that the complexes were organized as spherical and filamentous assemblies (Figures 1 and 2). This mode of organization of the cationic liposome/DNA complex is in agreement with the previously suggested “spaghetti-meatball” model that is based on freeze-fracture EM observations. “Meatball” represents liposomes, whereas “spaghetti” structures represent DNA filaments surrounded by cationic lipid bilayers (Sternberg et al. 1994). However, we report here for the first time that these structures have been imaged inside the cells, indicating that both structures (“spaghetti” and “meatball”) enter the cell. Which structure is the most efficient in catalyzing further steps of transfection still requires further investigation.
In summary, the BrdU labeling technique described here offers several advantages for exogenous DNA detection in the cells. First, the DNA is not significantly modified because BrdU is a nucleotide analogue. Second, by selecting the appropriate secondary antibody (gold or fluorescent conjugates), both electron and confocal microscopy can be applied to visualize the exogenous DNA inside the cells. This approach could easily be extended to other gene transfer methods and would allow better understanding of DNA trafficking from the cell surface to the nucleus during the transfection process.
Footnotes
Acknowledgements
M. Vandenbranden and M. Thiry are FNRS Research Associates. A. El Ouahabi obtained financial support from the David and Alice Van Buuren foundation.