
Abstract
Immunostaining techniques are commonly employed to determine the temporal and spatial patterns of cellular components in in situ preparations of cells and tissues. Usually, cells are formalin-fixed, permeabilized with nonionic detergents, and probed with specific antibodies. The incorporation of a sodium dodecyl sulfate (SDS) treatment after chemical crosslinking has been shown to improve the immunodetection of some cytosolic and cell surface antigens. By incorporating an SDS treatment after crosslinking, we report a significant improvement in the detection of two nuclear antigens (i.e., the DNA binding proteins apurinic/apyrimidinic endonuclease and DNA polymerase-β) and bromodeoxyuridine-tagged DNA by indirect immunofluorescence of whole cells. In bromodeoxyuridine-tagged DNA, the improvement in detection after an SDS treatment was observed only after long incorporation protocols (>48 hr) and, interestingly, it was more pronounced in cultured human foreskin keratinocytes than in bovine aorta endothelial cells. In addition, the SDS treatment proved in these studies to be superior to the standard Triton X-100 permeabilization. SDS thus provides a potential means to visualize previously undetectable or poorly detectable nuclear antigens. (J Histochem Cytochem 47:1095–1100, 1999)
Immunohisto/cytochemical techniques are used routinely to dissect the biological contribution(s) of cellular components and/or to determine protein expression patterns. However, in some instances these procedures do not permit suitable detection of specific antigens due to “masked” epitopes (Soltys and Gupta 1992; Ding et al. 1995; Brown et al. 1996; Robinson et al. 1996; Werner et al. 1996; Shi et al. 1997). Several methods to reveal cellular antigens, including microwaving (Werner et al. 1996; Shi et al. 1997), permeabilization with nonionic detergents (Robinson et al. 1996) and specifically, sodium dodecyl sulfate (SDS) treatment (Soltys and Gupta 1992; Ding et al. 1995; Brown et al. 1996; Bianchi et al. 1999) after chemical crosslinking have recently been reported. An SDS treatment permitted the detection of some cytosolic and cell-surface associated antigens. We describe here the use of an SDS treatment to detect two nuclear mammalian DNA repair proteins, apurinic/apyrimidinic endonuclease (APE) (Demple et al. 1991; Robson et al. 1991; Seki et al. 1992) and DNA polymerase-β (β-pol) (Wilson and Thompson 1997 and references therein) as well as bromodeoxyuridine (BrdU)-labeled (Bosq and Bourhis 1997; Ormerod 1997) bovine aorta endothelial (BAE) cells and human foreskin keratinocytes (HFKs).
APE and β-pol are central players in the multistep base excision DNA repair (BER) pathway, which is typically initiated by enzymes (DNA glycosylases) that catalyze the release of inappropriate bases from DNA (Wilson and Thompson 1997). APE (also known as Hap1, Ref-1, and APEX) is the predominant mammalian apurinic/apyrimidinic (AP) endonuclease and commences repair of spontaneously or enzymatically derived abasic sites in DNA by incising immediately 5′ to the lesion (Wilson et al. 1995a,b). APE also modulates the DNA binding activity of several AP-1 transcription factors (e.g., Fos, Jun, and NF-κB) in vitro through a redox mechanism that has been termed its Ref-1 function (Xanthoudakis et al. 1992). β-pol catalyzes the removal of the 5′ abasic residue remaining after AP endonuclease incision and also performs the synthetic gap-filling step of the BER process before DNA ligation (Singhal et al. 1995). The importance of BER is highlighted by the fact that mice lacking either APE/Ref-1 or β-pol do not survive embryogenesis (Sobol et al. 1996; Xanthoudakis et al. 1996). Therefore, BER likely protects against the lethal accumulation of naturally occurring DNA damage, such as alkylated and oxidized bases, AP sites, and strand breaks.
BrdU is a common marker to detect chromosomal DNA synthesis as an indicator of cell proliferation, and antibodies against BrdU are readily available (Bosq and Bourhis 1997).
Immunodetection analysis was undertaken to study the expression patterns of these two repair proteins as well as BrdU incorporation into chromosomal DNA. Initial immunofluorescence experiments revealed that the antibodies used against APE or β-pol exhibited nonspecific reactivity. However, an SDS step dramatically improved the ability of these polyclonal antibodies to recognize their target antigens in both HFK and BAE cells. In addition, an SDS treatment enhanced detection of BrdU-labeled DNA when BrdU was incorporated for long periods into both HFK and BAE cells. This improvement was more dramatic in HFKs. Implementation of an SDS treatment step may allow investigators to visualize previously undetectable nuclear proteins as well as DNA biomarkers (e.g., apoptosis-specific markers, DNA adducts) and may improve such techniques as in situ chromosomal hybridization (Hendzel and Bazett-Jones 1997) and TUNEL (Negoescu et al. 1996).
Materials and Methods
Immunoblotting
Total cell extracts were prepared from BAE cells or HFKs grown in DMEM (Mediatech; Hendon, VA) plus 10% bovine calf serum (CS) (Hyclone Laboratories; Logan, UT) and KGM Bulletkit (Clonetics; Walksville, CA), respectively. Cells were scraped from T75 flasks, collected by centrifugation, and lysed in Laemmli's buffer (62.5 mM Tris-HCl, 2% SDS, 1% β-mercaptoethanol, 10% glycerol, and 0.0025% bromophenol blue) (Laemmli 1970). The lysates were boiled for 5 min and 20 μl (#10 μg) was subjected to 10% SDS-PAGE. The proteins were transferred to Immobilon-P membranes (Millipore; Bedford, MA) using a semidry apparatus (BioRad; Richmond, CA). The membranes were blocked with 5% nonfat dry milk in TBST (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.05% Tween-20) for 1 hr at room temperature (RT). Blots were probed with antiserum to APE (#1397) (from Dr. Hussain) at 1:3000 dilution or β-pol (from Dr. Linn) at 1:2000 dilution in TBST plus 5% milk overnight at 4C. Unbound antibodies were removed by three 5-min washes in TBST, followed by incubation with anti-rabbit-F(ab)'2 coupled to alkaline phosphatase (Jackson Immunoresearch Laboratories; West Grove, PA) at 1:3000 dilution for 30 min at RT. Excess anti-rabbit-F(ab)'2 antibodies were removed as above and alkaline phosphatase was visualized colorimetrically using 5-bromo-4-chloro-3-indolyl-phosphate and nitroblue tetrazolium phosphatase substrate system (Kirkegaard & Perry Laboratories; Gaithersburg, MD). When enhanced chemiluminescence visualization was used, the secondary antibody was an anti-rabbit antibody coupled to horseradish peroxidase (1:2000) (Amersham; Little Chalfont, UK) and was processed as described in the manufacturer's instructions. In immunoabsorption experiments, antibodies were incubated with either 20 μg/ml of glutathione S-transferase-APE fusion protein (GST-APE) (Wilson et al. 1995b), GST-APE, or buffer alone for 1 hr at RT before incubation with the blots. Coommassie blue (0.1%) staining of the membrane was performed for 2 min, followed by a 10-min wash with 50% methanol/7% acetic acid solution at the end of the immunoblotting experiments to verify proper fractionation, transfer, and even protein loading.
Cell Manipulations
BAE cells and HFKs were cultured as described above. Cells were plated at 50% confluence on 1.5% gelatin-coated glass 8-well chambers (Nunc; Naperville, IL) and allowed to grow to confluence. In some cases, a “wound” was introduced by scraping away the cells in the middle of the chamber with a 1000-μl. pipette tip. The chambers were washed twice in fresh medium to remove unattached cells and the remaining cells were allowed to recover as indicated (see Figure legends).
BrdU Labeling
BrdU (Boehringer Mannheim; Indianapolis, IN) was added to the medium at the recommended concentration during the “wound” recovery for various time intervals (see Results) or for the last hour before fixation. BrdU incorporation was visualized according to the manufacturer's instructions unless otherwise indicated (see figure legends).
Indirect Immunofluorescence
All steps were performed at RT. Medium was aspirated from cells, and the chamber wells were washed once with PBS (0.9% NaCl in 10 mM sodium phosphate buffer, pH 7.6). Cells were fixed for 20 min in freshly made 4% formaldehyde (from paraformaldehyde) in PBS and then washed five times with PBS. At this stage, the cells were incubated for 5 min in either PBS alone or PBS containing 1% SDS (Sigma; St Louis, MO). In some experiments, cells were incubated with 0.5% Triton X-100 (Fisher Scientific; Pittsburgh, PA) for 5 min. All incubations were followed by five washes with PBS and blocking with PBS plus 1% BSA (Fraction V) and 0.05% saponin (both from Sigma) (blocking buffer) for 1 hr. Primary antibodies were then added to the chamber wells at dilutions of 1:3000 (APE) and 1:2000 (β-pol) in blocking buffer for 1 hr. This was followed by three washes with PBS and incubation with goat anti-rabbit IgG coupled to Cy3 (Sigma) at 1:500 dilution in blocking buffer for 30 min. Excess secondary antibody was removed by washing with PBS five times, and the chambers were then mounted in Vectashield medium (Vector Laboratories; Burlingame, CA). Immunofluorescence was visualized with an Olympus upright microscope equipped with a X40 oil immersion lens, the appropriate fluorescence filters and UV light sources (Molecular Medicine Unit; Beth Israel Deaconess Medical Center, Boston, MA). Pictures were recorded in Kodak TMAX 400 ASA at the same exposure time for all paired micrographs, and were developed and printed under identical conditions.
Immunoadsorption experiments with GST or GST-APE were performed as described earlier. Control experiments with the secondary antibody alone were routinely performed and produced no detectable immunofluorescence signal (not shown).
Results
Immunoblotting experiments revealed that antibodies raised against APE recognized primarily a single protein band of the expected size (Figure 1A) and that this band could be competed by an excess of recombinant GST-APE protein (Figure 1B). In immunofluorescence experiments, however, there was no detectable nuclear antigen staining (Figure 2A) and the signal was not inhibited by preincubation with GST-APE (Figure 2B). Introduction of an SDS treatment step after formaldehyde fixation of BAE cells produced the expected nuclear staining pattern with a lighter, albeit specific, cytosolic staining with the anti-APE antibodies (Figure 3A). Moreover, this immunofluorescence reaction was inhibited by recombinant GST-APE protein (Figure 3B) but not by GST alone (Figure 3C). Likewise, an SDS treatment of HFK produced a nuclear staining pattern that was APE-specific (not shown). Although similar immunoabsorption experiments were not performed with purified β-pol protein, the nuclear staining for this DNA polymerase was observed only after an SDS treatment (compare Figures 4A and 4B).
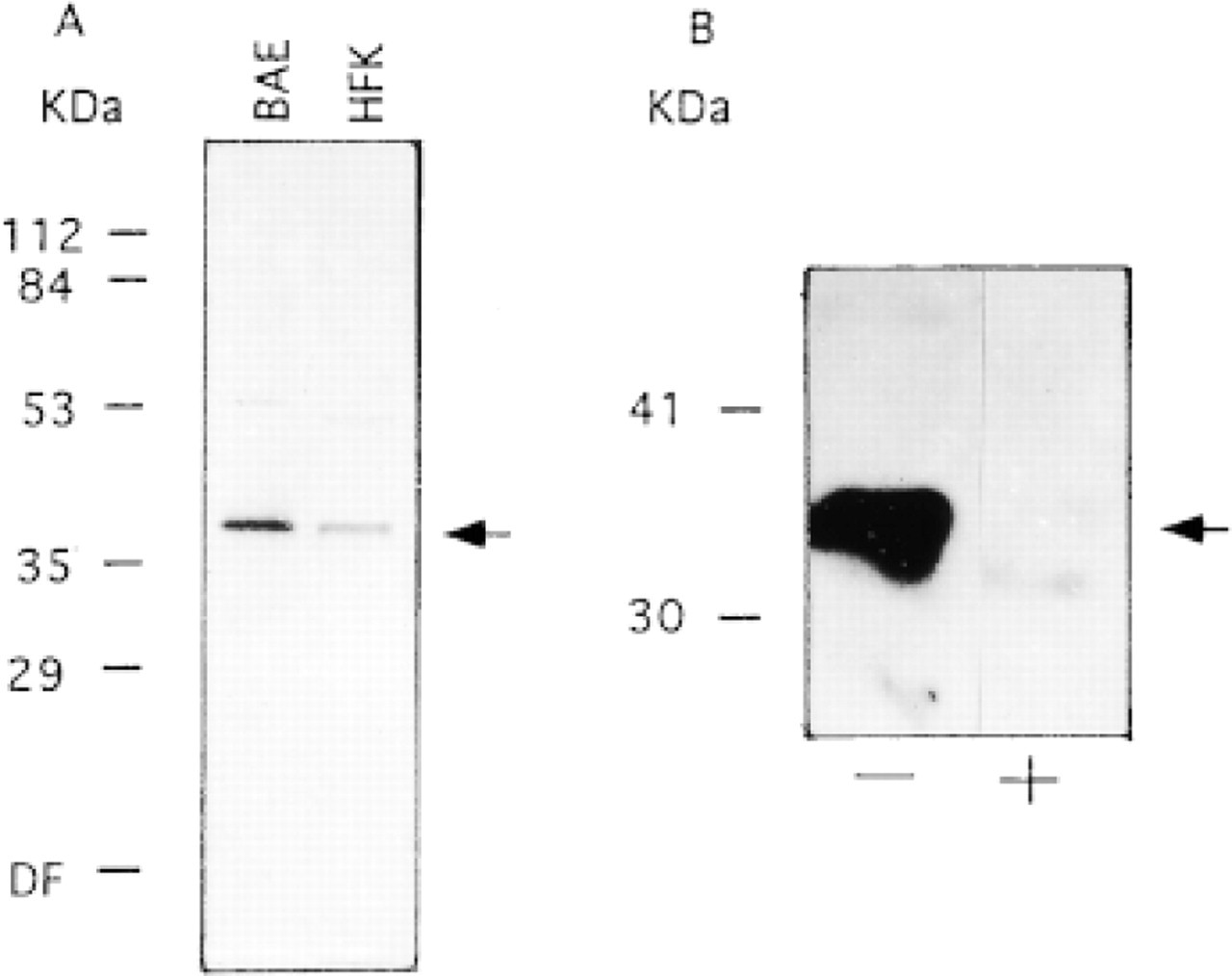
(A) Immunoblotting of APE (arrow) in bovine aorta endothelial (BAE) cells and human foreskin keratinocytes (HFKs) using antibody #1397. (B) The specificity of APE antibody recognition in BAE extracts without (−) or with (+) preabsorption with 20 μg/ml of purified recombinant GST-APE. Protein molecular weight (kD) markers are indicated.
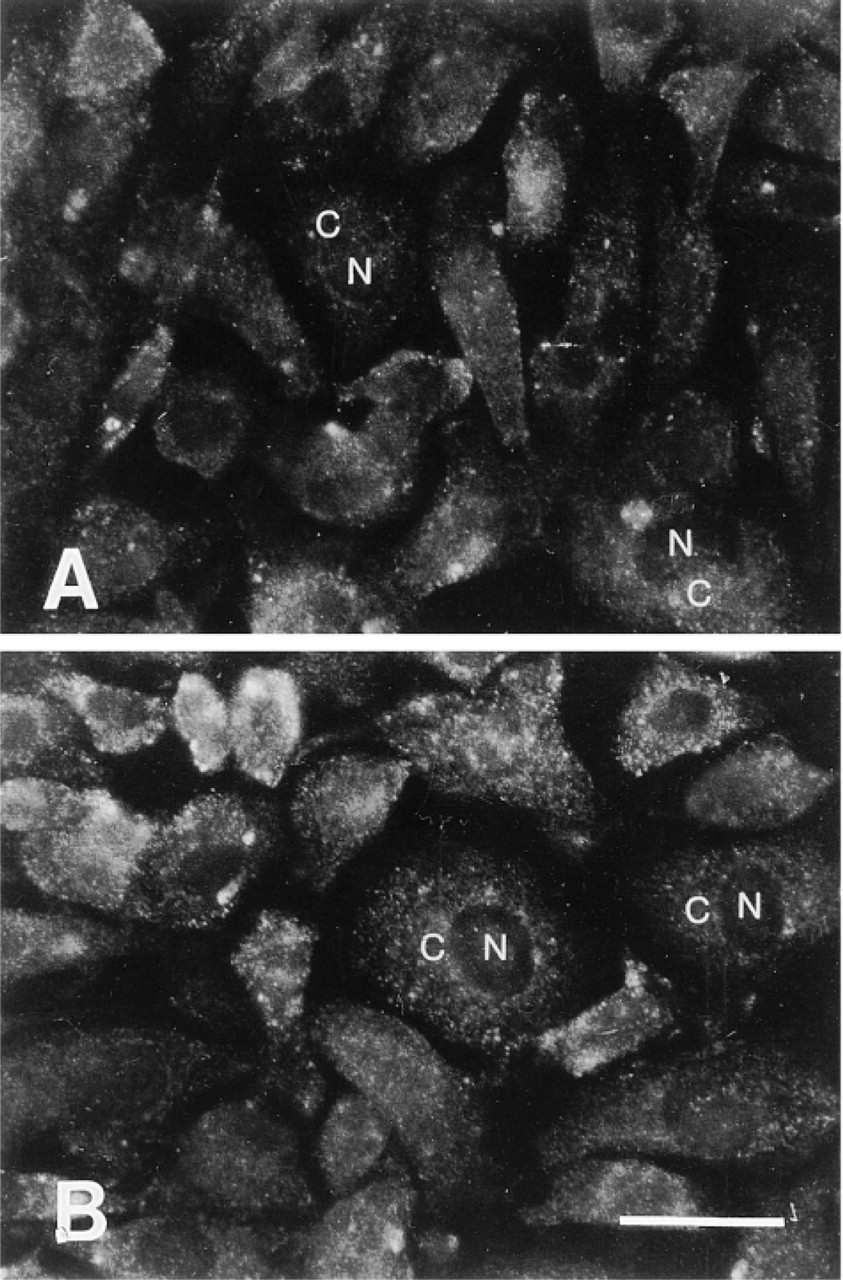
Indirect immunofluorescence without SDS treatment in BAE cells. Note the lack of specificity/localization of APE antibody either without (A) or with (B) preabsorption with 20 μg/ml of purified recombinant GST-APE. N, nuclei; C, cytoplasm. Bar = 10 μm.
To determine if APE expression patterns changed during the cell cycle, immunostaining experiments were performed on cells containing BrdU-tagged DNA. BrdU is incorporated into DNA during chromosomal replication. During these experiments, SDS treatment was found to enhance the labeling of BrdU in BAE cells (Figures 5A and 5B). This enhancement was observed when BrdU had been incorporated for >24 hr relative to freshly incorporated BrdU (<2 hr; not shown) and the enhancement of BrdU detection was even more dramatic in HFKs (not shown).
Discussion
Organisms possess a variety of DNA repair systems to maintain their genetic integrity in the face of endogenous and exogenous DNA-damaging agents. Studies of DNA repair mechanisms, including nucleotide excision repair, BER, mismatch repair, strand break repair, and recombination repair, are of great importance, because of the association of DNA repair defects with human disease, most notably cancer (Demple et al. 1997). One attractive way to characterize these systems, as well as other biological systems, is by using immunocyto/histochemistry techniques. No specific staining, however (nuclear or cytosolic), was detected in cultured cells using APE-specific antibodies in initial immunofluorescence studies (Figure 2A), and the immunofluorescence signal was not competed by GST-APE (Figure 2B). Therefore, approaches to improve antigen recognition and antibody specificity were investigated.
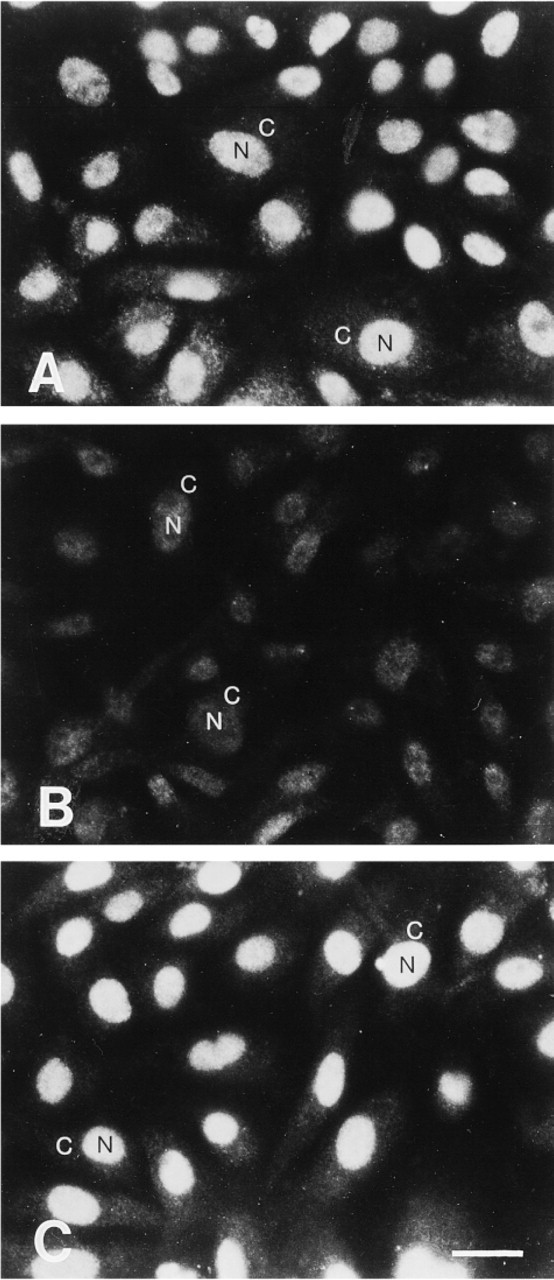
Indirect immunofluorescence with SDS treatment in BAE cells. Note the specific localization of staining of the APE #1397 antibody. The nuclear staining is observed when the antibody is used alone (A), decreases dramatically when preabsorbed with 20 μg/ml of purified recombinant GST-APE (B), but is not affected by pread-sorption with 20 μg/ml of purified recombinant GST (C). N, nuclei; C,cytoplasm. Bar = 10 μm.
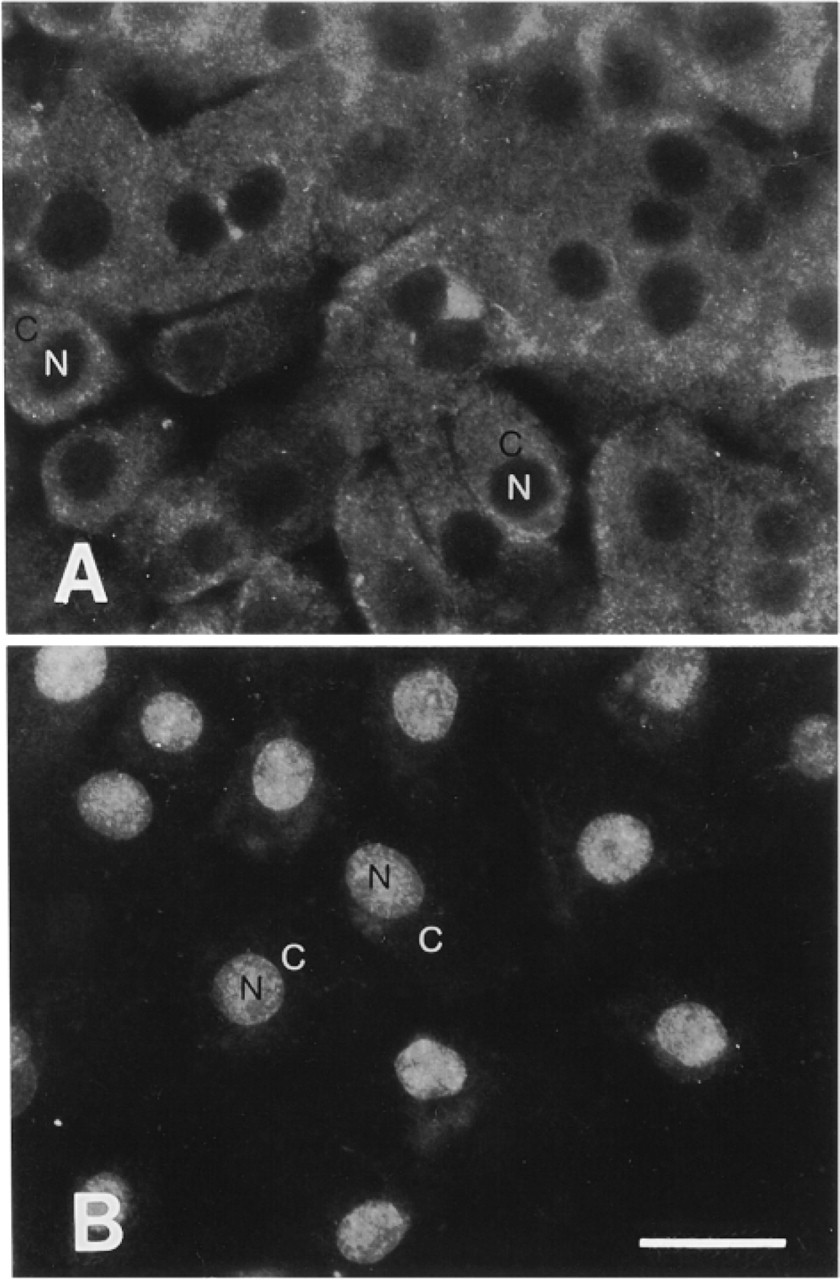
Indirect imunofluorescence illustrating the effect of SDS treatment in the detection of β-pol specific antibodies in HFKs, without SDS treatment (A) and with SDS treatment (B). N,nuclei; C,cytoplasm. Bar = 10 μm.
We incorporated an SDS treatment step (Soltys and Gupta 1992; Ding et al. 1995; Brown et al. 1996; Bianchi et al. 1999) in the hope of developing conditions that would permit the use of polyclonal antibodies raised against two nuclear proteins belonging to the BER pathway, i.e., APE and β-pol, in immuno-fluorescence studies. Previous studies demonstrated the use of an SDS to detect cytosolic and membrane-associated proteins in immunofluorescence experiments and we therefore attempted to extend this method to the detection of nuclear antigens.
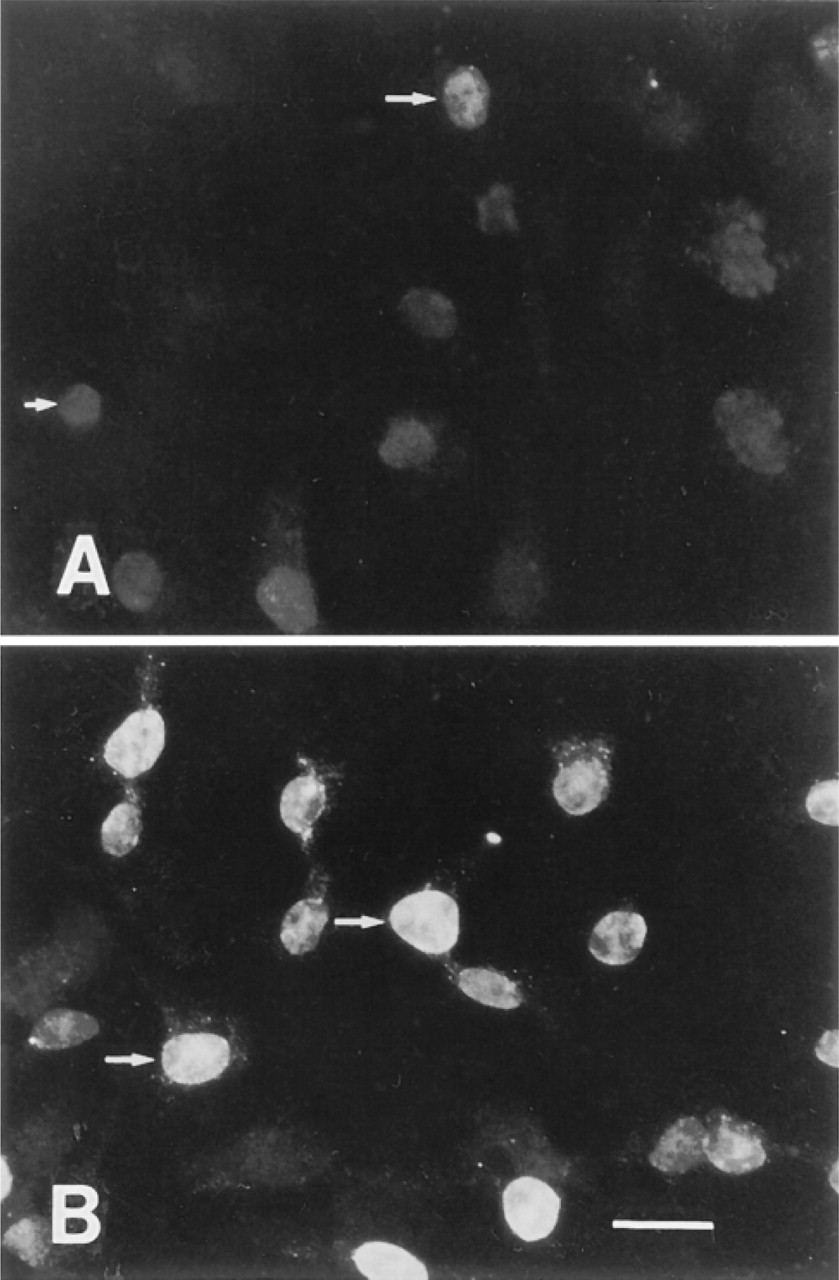
Immunofluorescence illustrating the effect of SDS treatment in the detection of BrdU-labeled DNA (>48 hr incorporation) in BAE cells using an anti-BrdU antibody. Immunofluorescence detection without SDS treatment (A) or with SDS treatment (B) of cells at the wound borders. Arrows, nuclei. Bar = 10 μm.
The improved antigen recognition after SDS treatment seen with both these antibody preparations may result from an increase in the permeability of the nuclear compartment and/or from exposure of the antigenic epitope(s) by either denaturation or dissociation of protein-protein or DNA-protein interactions.
It appears reasonable to predict that SDS treatment would function to dissociate protein-protein, DNA-DNA, and protein-DNA interactions which, in turn, would expose BrdU to antibody recognition. BrdU-labeled DNA that is associated with various auxiliary factors is likely to be more abundant after multiple cycles of BrdU incorporation (i.e., when BrdU was maintained in the medium for more than one doubling time, 24 hr), perhaps explaining the more dramatic increase in immunostaining under such conditions. When we compared SDS treatment with Triton X-100 permeabilization in BAE cells, we observed that both protocols were successful but that the SDS-treated cells produced a more intense signal (not shown).
These studies should stimulate investigators to examine the effects of SDS treatment in using BrdU labeling techniques and in analyzing the cell localization patterns of their target gene products. Finally, future experiments will indicate if SDS is useful for the unmasking (retrieval) of other antigens.
Footnotes
Acknowledgments
Supported in part by a National Research Service Award from the National Cancer Institute to DMW III (CA62845) and in part by 1 P50 HL56993-01 (SCOR in Molecular Medicine and Atherosclerosis) to CB.
We thank Drs S. Alper and A. Stuart-Tilley for introducing the SDS protocol and for the use of the fluorescence microscope; Drs B. Demple, F. W. Sellke, and B. G. Neel for their continuous encouragement and support; Dr. M. Karnovisky for critical review of the manuscript; and Dr R. Schlegel for HFK, Ms A. Hampson for excellent illustration work; and Drs S. Linn and I. Hussain for the antibodies against β-pol and APE, respectively.