
Abstract
It has been repeatedly shown that synaptically released zinc contributes to excitotoxic neuronal injury in ischemia, epilepsy, and mechanical head trauma. Such zinc-induced injury leaves an unmistakable “footprint” in the injured neurons, allowing an easy and unambiguous postmortem diagnosis. This footprint is the presence of weakly bound, histochemically reactive zinc in the cytoplasm of the perikaryon and proximal dendrites. Such staining appears to be a necessary and sufficient marker for zinc-induced neuronal injury. Here we show how to prepare and stain tissue from biopsy, autopsy, or experimental animal sources for maximal contrast and visibility of zinc-injured neurons.
D
As of this writing, the only way to diagnose prior zinc-induced neuronal injury in a biopsy or autopsy specimen is to stain the tissue for zinc and search for signs of zinc translocation. Evidence of prior zinc translocation epsiodes includes both depletion of zinc from presynaptic boutons and the anomalous appearance of weakly bound zinc in the cytoplasm of injured postsynaptic neurons. Of these two signs, the latter is by far the most reliable because normal healthy neurons in the brain never show any staining for zinc in the perikaryon or proximal dendrites. In general, therefore, the appearance of the atypical, anomalous zinc staining in the perikaryon is both a necessary and a sufficient condition for the diagnosis of a zinc translocation-mediated neuronal injury.
The present studies were undertaken in search of optimal zinc staining methods for detecting zinc translocation. Both fluorescent and silver-amplification histochemistry were explored and a number of variations in tissue preparation and staining methods were tested. Special emphasis was placed on the use of these methods to identify zinc translocation and thus to diagnose zinc-induced neuronal injury in tissue that was taken from human subjects as autopsy or biopsy specimens.
Materials and Methods
Fluorescent probes included TSQ (Frederickson et al. 1987, 1992) and its congener TFlZn (Teflabs; Austin TX) and an enzyme-based system employing zinc-free human carbonic anhydrase II (apoCA) as the “detector” with the fluorochrome ABD-N (7-(N-amino ethane-2-ol)benz-2-oxa-1,3-diazole-4-sulfonamid) as the “reporter” [see Thompson et al. (1997) for further methods]. The silver method was the frozen-tissue, sulfide gas method recently developed by Danscher and colleagues for human autopsy/biopsy material [see Danscher et al. (1997) for details].
Several models of zinc-induced neuronal injury were explored, including kainic acid (KA)-induced seizures [Methods in Frederickson et al. (1988, 1989)] and mechanical brain trauma (Long et al. 1998). In addition, some brain slices [prepared and maintained as in Howell et al. (1984) and Easley et al. 1995)] and hippocampal organotypic cultures (Whetsell and Schwarcz 1983) were subjected to excitotoxic insults in vitro and stained either in vitro or after harvesting, freezing, and sectioning. For the acute slices, simply interrupting the superfusate flow for 1 hr was the insult. For the cultures, the insult was to delay supplying fresh medium and oxygen for 1-2 days [see Whetsell and Schwarcz (1983) for details of this ischemia/hypoglycemia method]. In the KA model of zinc-induced cell death (Frederickson et al. 1988, 1989), the KA was administered
Results
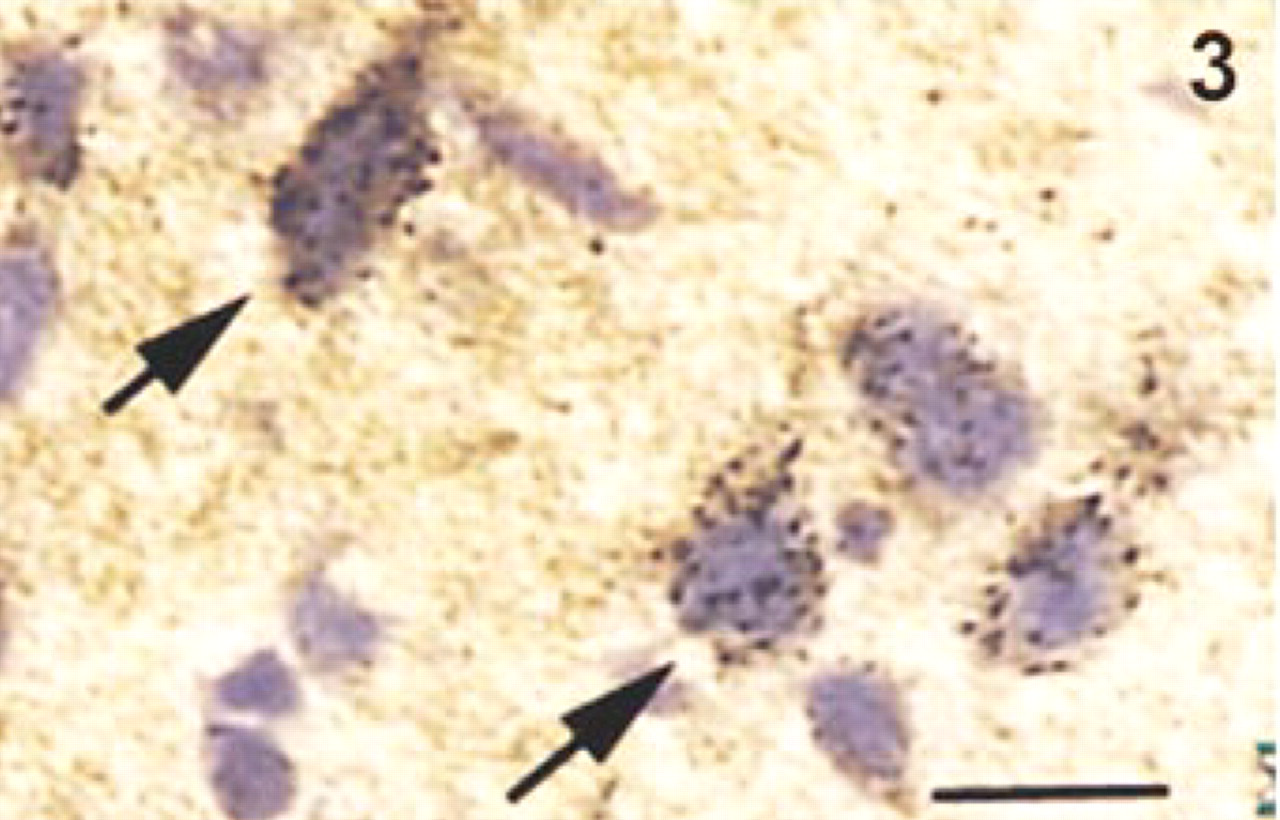
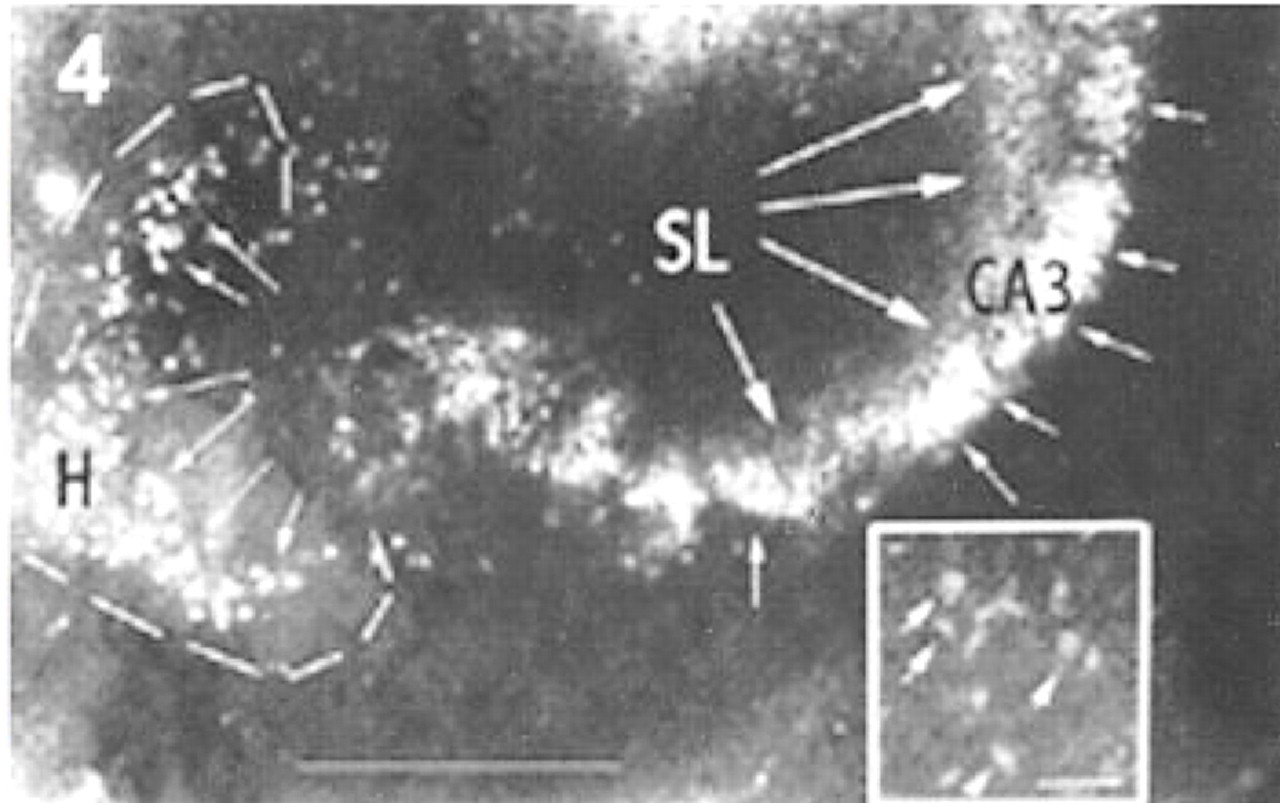
Five results merit presentation. First, as has been shown previously for bouton zinc staining (Frederickson et al. 1987) the time elapsed between death (or live tissue harvesting) and the freezing of the tissue has proved to be critical. Tissue that has remained unfrozen for more than 1-2 hr post mortem shows an arti-factual decline in the intensity of fluorescent staining that declines to virtually no staining at all by 12-24 hr. Likewise, in normal tissue, the staining of boutons declines over 12-24 hr and there is no corresponding increase in the staining of neuronal perikarya (not shown). Second, the manner in which the tissue is frozen can determine whether or not neurons are found. Specifically, freezing procedures that promote morphological damage, such as freezing too slowly or too rapidly, or (worse) freezing, thawing, and refreezing, can obliterate the discernible staining of zinc-injured neurons (Figure 1). The third important finding is that a histological marker that cannot easily penetrate cell membranes is superior for highest-contrast definition of zinc-injured neurons. This is shown in Figure 2, in which the staining of CA3 pyramidal neurons (injured by KA-induced status epilepticus) is vivid, whereas the adjacent mossy fiber neuropil is virtually unstained. The failure of staining in the neuropil, coupled with the positive reaction in the perikarya, enables the latter to stand out in high contrast. The fourth noteworthy finding is that zinc-injured neurons can be reliably and densely stained in autopsy or biopsy material by a variation of the sulfide gas-silver method of Danscher et al. (1997). The variation that allows the injured perikarya to be stained (in addition to the normal bouton staining) is to treat the cut tissue sections with a basic (pH 10) buffer (so called “universal buffer” (Na-barbital/Na-acetate) (Frederickson et al. 1987) before exposing them to the sulfide gas. For this variant, one immerses the cut and mounted cryostat sections in the buffer for 2 min, rinses briefly in saline, then places the tissue in the GAS-AMG exposure chamber, as described by Danscher et al. (1997). Whereas few zinc-killed neurons can be stained by conventional silver methods for zinc (Sloviter, 1985; Frederickson et al. 1989), many (perhaps all) are stained by this novel variant (Figure 3). Fifth, and finally, neurons subjected to ischemic insult in vitro, as cut slices or organotypic cultures, can be readily stained by a cell-permeable fluorescent marker, in vitro or after frozen sectioning, by the standard TSQ methods (Figure 4).
Discussion
The present results show that the postmortem diagnosis of zinc-mediated neuronal injury can be carried out on any tissue that has been (a) harvested within 1-2 hr of death, (b) left unfixed, and (c) frozen properly and maintained frozen. Such tissue can be stained immediately with either the TSQ (Frederickson et al. 1987) or the CA-ABDN method (Thompson et al. 1997), photographed, then postfixed for archival staining. Alternatively, one can treat the sections with basic buffer, then use the sulfide gas method for permanent light-and electron-dense silver staining of tissue (Danscher et al. 1997). In all cases, thinner (10-μm) sections give better cell definition than thicker.
The reason that a short postmortem period is critical is presumably that the Zn2+ present in the fresh tissue binds fairly rapidly with protein residues and small molecules (including S−) liberated by postmortem autolysis. Concerning the peculiar sensitivity of the zinc-positive cell staining to freeze-thaw-freeze damage (freezer burn), we speculate that this is because the Zn2+ (unlike the macromolecules of Nissl) is a mobile ion in solution and would be expected to diffuse quickly away through ruptured membranes. The “smudges” or “clouds” of fluorescence staining that remain after such diffusion are difficult to recognize as former neurons.
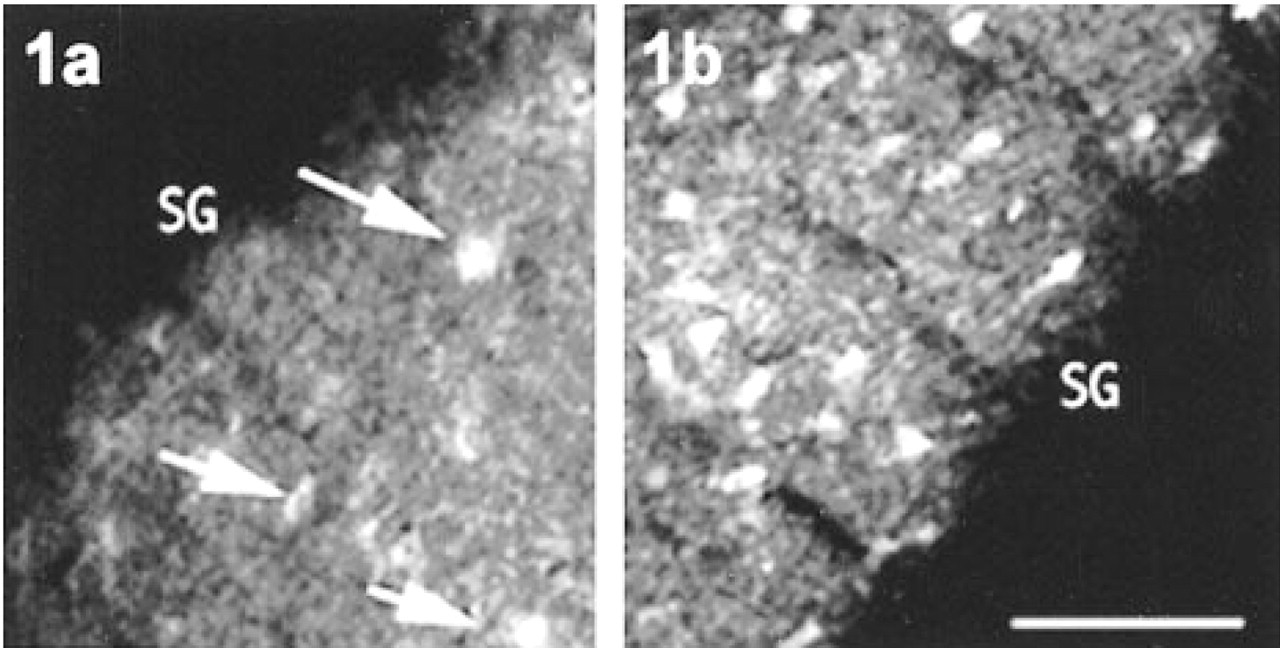
Effect of refreezing on nearby sections of rat dentate gyrus hilar region. This rat was subjected to 3 hr of kainic acid-induced convulsions before sacrifice, resulting in zinc translocation into somata. Standard TSQ methods were used to stain both sections. (

Staining of neuronal somata by carbonic anhydrase-based zinc stain. After 4 hr of KA-induced convulsions, this rat was sacrificed and the brain was cut frozen, thawed, and stained. The stain was a mixture of 1 μm zinc-free human carbonic anhydrase and the fluorochrome ABDN (Thompson et al. 1997), which fluoresces brightly (560-nm emission, 430-nm excitation) when the carbonic anhydrase binds any available free zinc. Many pyramidal somata in stratum pyramidale (SP) are vividly fluorescent, whereas the mossy boutons in stratum lucidum (SL) are not appreciably brighter than the subjacent stratum oriens (SO). Bar = 50 μm.
Silver staining for translocated zinc. This tissue was harvested from a rat (dorsal cortex) subjected to traumatic head injury, which causes translocation of zinc from boutons to somata. The silver grains were produced by the Danscher gas-AMG method, in which the fresh-frozen, unfixed sections are exposed to sulfide gas to create ZnS catalyst, then developed by the Danscher AMG development (Danscher et al. 1997). This silver-stained material would be suitable for EM determination of the exact localization of the translocated zinc. Bar = 30 μm.
TSQ staining of translocated zinc in neuronal somata of hippocampal culture and (
The advantage of CA-ABDN as a stain for zinc-filled neurons was an unexpected finding, which we tentatively interpret thus. If most of the vesicles in zinc-containing boutons are intact after single freezing and cutting of the tissue at 10-20 μm, then most of the bouton zinc (in the vesicles) would be unstained by the membrane-impermeable CA-ABDN. Most neural somata, on the other hand, will be sliced open (especially in the thinner 10 μm sections) and thus open to the CA-ABDN. Alternatively, it is possible that the biochemical microenvironment of the zinc in vesicles is sufficiently different from that of the zinc in the perikarya that the CA-ABDN staining favors the latter. This idea is also consistent with the finding that pretreatment with a basic buffer is needed to achieve consistent staining of perikaryal zinc, but not vesicular zinc, with the sulfide gas method.
We know of only three conditions in which neurons in the brain will show staining for zinc: after excitotoxic zinc translocation (as in this report), in Alzheimer's disease (Suh et al. 1998), and after exposure to a pathological excess of NO (Cuajungco and Lees 1998). In Alzheimer's, there is evidence that excess synaptically released zinc may be a contributing factor in the neuronal pathology. In the case of the NO-mediated injury, it is also quite plausible that the NO causes release of zinc from vesicles, thus triggering the familiar zinc translocation cell injury.
Zinc translocation in stroke, ischemia, hypoxia, seizures, and head injury may be a leading cause of neuronal injury in adults. The ability to identify and diagnose this particular pathology in any suitably collected and archived tissue sample should facilitate further research.
Footnotes
Acknowledgements
Supported in part by NIH-GM 48894, MH 56335, and NS 37658 (to CJF), by the Schriners Burn Foundation (JWC), NSF (RBT, DSS), the Danish Medical Research Council (SWS, GD), and the Office of Naval Research (RBT).
We thank Gabrielle Schneider for technical assistance.