
Abstract
We investigated the enzymes involved in the NADPH-diaphorase (d) reaction in the rat and pig bladder urothelium. The urothelial cell layer displayed intense and uniform NADPH-d activity. Preincubation with the flavoprotein inhibitor diphenyleneiodionium chloride (DPI) and the alkaline phosphatase inhibitor levamisole concentration-dependently decreased the urothelial NADPH-d activity. Immunoreactivities to neuronal (n), endothelial (e), or inducible (i) nitric oxide synthase (NOS) were not detected in rat or pig urothelial cells. In rats, the urothelium was uniformly immunoreactive for NADPH cytochrome P450 reductase, whereas the pig urothelium displayed inconsistent labeling. In lipopolysaccharide (LPS)-treated rats, the bladder urothelium showed positive iNOS immunoreactivity. The iNOS labeling was found predominantly in cells located in the basal layer of the urothelium. In the pig bladder mucosa, a Ca2+ -dependent NOS activity was evident in cytosolic and particulate fractions that was quantitatively comparable to the NOS activity found in the smooth muscle. In ultrastructural studies of urothelial cells, NADPH-d reaction products were found predominantly on membranes of the nuclear envelope, endoplasmatic reticulum and mitochondria. In conclusion, NADPH-d staining of the urothelium cannot be taken as an indicator for the presence of constitutively expressed NOS. Activity of alkaline phosphatase and cytochrome P450 reductase may account for part of the NADPH-d reaction in urothelial cells. However, LPS treatment of rats caused expression of iNOS in urothelial cells.
Keywords
N
There is now considerable evidence that NADPH-d activity stains cells that lack NOS and that NADPH-d histochemistry cannot be considered an accurate marker for the presence of NOS (Beesley 1995; Blottner et al. 1995). This histochemical technique relies on a simple redox reaction which merely depends on NADPH and electron transfer to nitroblue tetrazolium. NADPH-d staining may have limited relation to NOS because several other enzymes, such as NADPH cytochrome P450 reductase, NADPH oxidase, and nonspecific phosphatases, may produce a similar reaction (Blottner et al. 1995).
The aim of the present study was to investigate which enzyme(s) is involved in the NADPH-d activity in the rat and pig urothelium. The subcellular localization and distribution of NADPH-d staining were investigated by electron microscopy to gain further insight into the characteristics of the NADPH-d staining in urothelial cells. In addition, a biochemical measurement of NOS activity was performed by using the conversion of radiolabeled L-arginine to L-citrulline (Bredt and Snyder 1989). This latter method detects all NOS activity in the tissue and therefore is not limited to the specificity of antisera. A preliminary report including some of these findings has been presented previously (Persson et al. 1997).
Materials and Methods
Tissue Handling
Female Sprague-Dawley rats (body weight 200-250 g) were sacrificed by CO2 asphyxia and the bladder was removed. Bladders from female pigs were obtained at a local slaughterhouse.
To induce NOS in vivo, some rats were given
To stimulate synthesis of cGMP, bladder pieces were placed in PBS at 37C and bubbled with a mixture of 95% O2/5% CO2 as previously described (Smet et al. 1996). To prevent the breakdown of cGMP, the buffer contained the phosphodiesterase inhibitors 3-isobutyl-1-methyl-xanthine (IBMX, 1 mM) and zaprinast (0.1 mM). After a 30-min incubation period with IBMX (Sigma) and zaprinast (May & Baker; Dagenham, UK), the tissue was stimulated by 1 mM sodium nitroprusside (SNP, Nipride; Roche, Basel, Switzerland) for 10 min. Control bladders were not exposed to SNP. The bladders were fixed in cold 4% formaldehyde in PBS for 4 hr and, for stimulated tissue, SNP also was included in the fixative.
NADPH-diaphorase Histochemistry
Tissue sections were preincubated in 50 mM Tris-HCl buffer for 10 min at room temperature (RT) and then incubated with 1 mM β-NADPH and 0.5 mM nitroblue tetrazolium (both from Sigma) dissolved in 50 mM Tris-HCl buffer containing 0.2% Triton X-100 for 90 min at 37C. Control experiments were performed without addition of β-NADPH. After rinsing in PBS, the sections were mounted in Kaiser's glycerol gelatin (Merck; Darmstadt, Germany). The effects of the flavoprotein inhibitor DPI (10-50 μM; Sigma) and the alkaline phosphatase inhibitor levamisole (5-10 mM; Sigma) on the NADPH-d staining were investigated after preincubation of the sections with the inhibitors for 30 min.
Immunohistochemistry
The tissues were immersion-fixed for 4 hr in cold 4% formaldehyde in 0.1 M PBS and then rinsed in PBS containing 15% sucrose for 2-3 days. Both fixation and rinsing were performed at 4C, after which the specimens were frozen in isopenthane at −40C and stored at −70C before sectioning. Tissue sections were cut at a thickness of 10 μm and preincubated with PBS containing 0.25% Triton X-100 for 2 hr at RT. Incubation with primary antisera was performed overnight in the presence of one of the following antisera: rabbit antisera raised against a C-terminal fragment of nNOS (1:1500; Eurodiagnostica, Malmö, Sweden), an N-terminal fragment of nNOS (1:1000; Eurodiagnostica), iNOS (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), eNOS (1:1000; Santa Cruz Biotechnology), NADPH-cytochrome P450 reductase (1:1000; Stress Gen Biotechnologies, Victoria, BC, Canada), or mouse antisera raised against eNOS (1:500; Transduction Laboratories, Lexington, KY), cytokeratin 8.13 (1:200; Sigma), ED-1 (1:500; Serotec, Oxford, UK), or sheep antisera raised against cGMP (1:1000; a generous gift from Dr J. de Vente, Limburg University, Maastricht, Netherlands). The cGMP antiserum has been described in detail previously (de Vente et al. 1987). The primary antisera were diluted in PBS containing 1% bovine serum albumin (BSA) and 0.25% Triton X-100. For visualization of the immunoreactive products, the sections were rinsed in PBS and then incubated for 90 min with either FITC (fluorescein isothiocyanate)-conjugated donkey anti-rabbit IgG (1:80; Jackson Immunoresearch Laboratories, West Grove, PA), FITC-conjugated donkey anti-sheep IgG (1:80; Sigma), or Texas Red-conjugated affinity purified F(ab')2 fragments of donkey anti-mouse IgG (1:160; Jackson Immunoresearch) diluted in PBS containing 1% BSA. All incubations with primary and secondary antisera were performed at RT in moisture chambers. The sections were finally rinsed and mounted in PBS/glycerol with p-phenylenediamine to prevent fluorescence fading. An Olympus epifluorescence microscope equipped with appropriate filter settings for Texas Red and FITC immunofluorescence was used.
For double label immunofluorescence of iNOS and ED-1, sections were first incubated with iNOS antiserum overnight, rinsed in PBS, and then incubated with ED-1 antiserum. After rinsing, the sections were incubated for 90 min with FITC-conjugated donkey-anti rabbit IgG, rinsed, and then incubated for 90 min with Texas Red-conjugated donkey anti-mouse IgG. After rinsing, the sections were mounted as previously described. In control experiments, no immunoreactivity could be detected in sections incubated without primary antibodies. Because crossreactions with antigens sharing similar sequences cannot be excluded, the structures demonstrated are referred to as, e.g., NOS-immunoreactive.
Electron Microscopy
For visualization of NADPH staining at the electron microscopic level, the specimens were fixed with 4% paraformaldehyde and 1% glutaraldehyde in 0.1 M Sørensen's phosphate buffer (SPB, pH 7.2) for 4 hr at 4C. Several washes in SPB followed, after which the specimens were embedded in 3% agarose and sectioned at 150 μ m on a vibratome. Vibratome sections were incubated with 1 mM β-NADPH and 0.5 mM 2-[2'-benzothiazolyl]-5-styryl-3[4 '-phthalhydrazidyl]-tetrazolium chloride (BSPT; Sigma) in 0.05 M Tris-HCl buffer (pH 8.0). Incubation was performed in darkness for 3-4 hr at RT. After several washes in Tris-HCl buffer and 0.15 M sodium cacodylate buffer, the sections were postfixed for 1 hr at 4C in 1% osmium tetroxide in 0.15 M sodium cacodylate buffer. A dehydration through a graded series of ethanol followed, and the specimens were embedded, sectioned, and contrasted according to conventional electron microscopic procedures. All grids were viewed and examined with a JEOL 1200 EX electron microscope. Control experiments were performed in the presence of BSPT but without addition of β-NADPH.
Measurement of Nitric Oxide Synthase Activity
NOS activity was determined by measuring the formation of L-[14 C]-citrulline from L-[14 C]-arginine according to the procedure described by Garcia-Pascual et al. (1996), with slight modifications. This NOS assay requires a substantial amount of tissue and was therefore performed using pig bladders. The bladder wall was dissected into muscle specimens representing the smooth muscle layer and mucosal specimens representing primarily the urothelium.
Tissues were homogenized in ice-cold buffer using a glass pestle homogenizer. Homogenization buffer (pH 7.2) contained 20 mM HEPES, 0.5 mM EDTA, 1 mM DTT, 1 mM PMSF, 1 μM pepstatin A, and 2 μM leupeptin. Homogenates were centrifuged at 20,000 μg for 30 min at 4C and the resultant cytosolic fractions were passed over a column of AG50W-X8 (Na+ form, prepared from the H+ form) to remove endogenous arginine. The pellet was resuspended in homogenization buffer containing 1 M KCl for 5 min to remove loosely bound cytosolic proteins and after centrifugation (20,000 × g for 20 min at 4C) the particulate fraction was resuspended in homogenization buffer. NOS activity was determined in the cytosolic and the KCl-washed particulate fractions. Incubation mixtures contained 75 μ l of cytosolic or particulate fractions and 25 μl of homogenization buffer containing, 2 mM NADPH, 1 mM CaCl2, 30 U/ml calmodulin, 3 μM tetrahydro-L-biopterin (BH4), 40 μ M L-arginine, and 0.8 μM L-[14 C]-arginine. Samples were incubated for 45 min at 37C in a shaking water bath. Preliminary experiments showed that the reaction was linear during this time. Incubation was terminated by the addition of 1.5 ml ice-cold stop buffer (5 mM HEPES, 2 mM EDTA, pH 5.5). Samples were passed through a 1-ml column of AG50W-X8 and eluted with 2 ml of stop buffer. Radioactivity of the effluent was measured by liquid scintillation spectrometry. A parallel set of incubations was performed on ice and was considered as nonspecific activity. To examine Ca2+ dependence, some experiments were performed in the absence of CaCl2 and calmodulin but in the presence of 2 mM EGTA. The activity in the presence of the NOS inhibitor L-NOARG (0.1 mM) also was studied. Ca2+ independent activity was determined as the difference between samples containing EGTA and samples containing L-NOARG. Ca2+ -dependent activity was calculated by subtracting the Ca2+ -independent activity from total NOS activity. Protein concentrations were determined according to Bradford using bovine serum albumin as standard. All measurements were made in duplicate and the specific activity was expressed as pmol citrulline/mg protein/min.
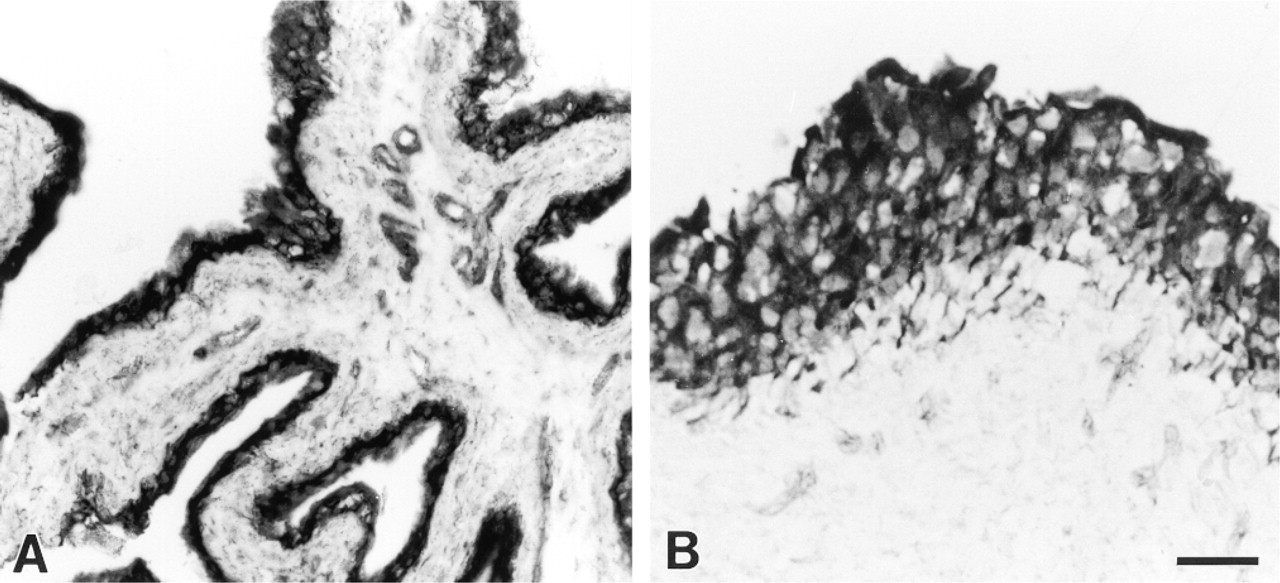
Histochemical demonstration of NADPH-d activity in the rat (
Results
Histochemical Demonstration of NADPH-diaphorase
Histochemical demonstration of NADPH-d showed a solid blue reaction product. The urothelial cell layer in rats and pigs displayed an intense and uniform NADPH-d activity (Figures 1A and 1B). Nerve fibers and endothelial cells were also stained. The urothelial NADPH-d reaction was further characterized in the rat bladder. The presence of the flavoprotein inhibitor DPI (10-50 μ?) and the alkaline phosphatase inhibitor levamisole (5-10 mM) concentration-dependently decreased the intensity of the urothelial NADPH-d labeling (Figure 2). Preincubation with levamisole decreased the intensity of the urothelial NADPH-d reaction, whereas levamisole seemed to have no effect on the neuronal and endothelial NADPH-d staining (Figure 2B). At the highest concentration of DPI, weak NADPH-d staining persisted in the urothelium, whereas all neuronal and endothelial staining seemed to have disappeared (Figure 2C).
Immunohistochemical Investigation of NOS and Related Enzymes
Cytokeratin 8.13. The anti-cytokeratin 8.13 antiserum, which reacts with a wide variety of epithelial cells (Gigi-Leitner et al. 1986), was used to define the urothelium. The urothelial cell layer showed distinct and uniform cytoplasmatic cytokeratin 8.13 immunoreactivity in both the rat and pig bladders (Figures 3A and 3B).
nNOS and eNOS. The C- and N-terminal antisera against nNOS labeled nerve fibers in the rat and pig bladders. However, no nNOS labeling was seen in either rat or pig urothelial cells (Figures 4A and 4B). A polyclonal antibody against eNOS was used in the rat bladder after confirmation of endothelial cell staining, while a monoclonal antibody was found to give a more reliable staining of endothelial cells in the pig bladder. However, no immunoreactivity to eNOS was found in the pig or rat urothelium (Figures 4C and 4D).
NADPH Cytochrome P450 Reductase. In rats, the urothelium was uniformly immunoreactive for cytochrome P450 reductase (Figure 3C). In addition, some endothelial cells occasionally showed immunoreactivity to cytochrome P450 reductase. Although the majority of the pig bladders investigated showed no urothelial cytochrome P450 reductase immunoreactivity, a low-intensity staining of urothelial cells was detected in some specimens.
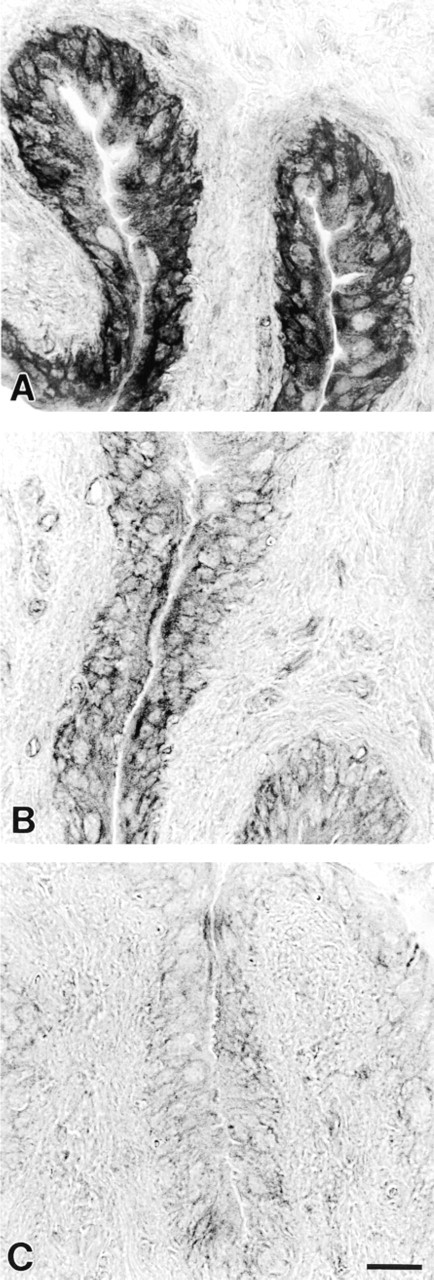
Histochemical demonstration of NADPH-d activity in the rat urothelium. (
cGMP. No immunoreactivity to cGMP was detected in the urothelium of SNP-stimulated rat bladders. However, in SNP-stimulated bladders the endothelium and the smooth muscle of intramural vessels showed intense cGMP immunoreactivity (not shown). Unstimulated control preparations did not show any cGMP immunoreactivity in vascular structures.
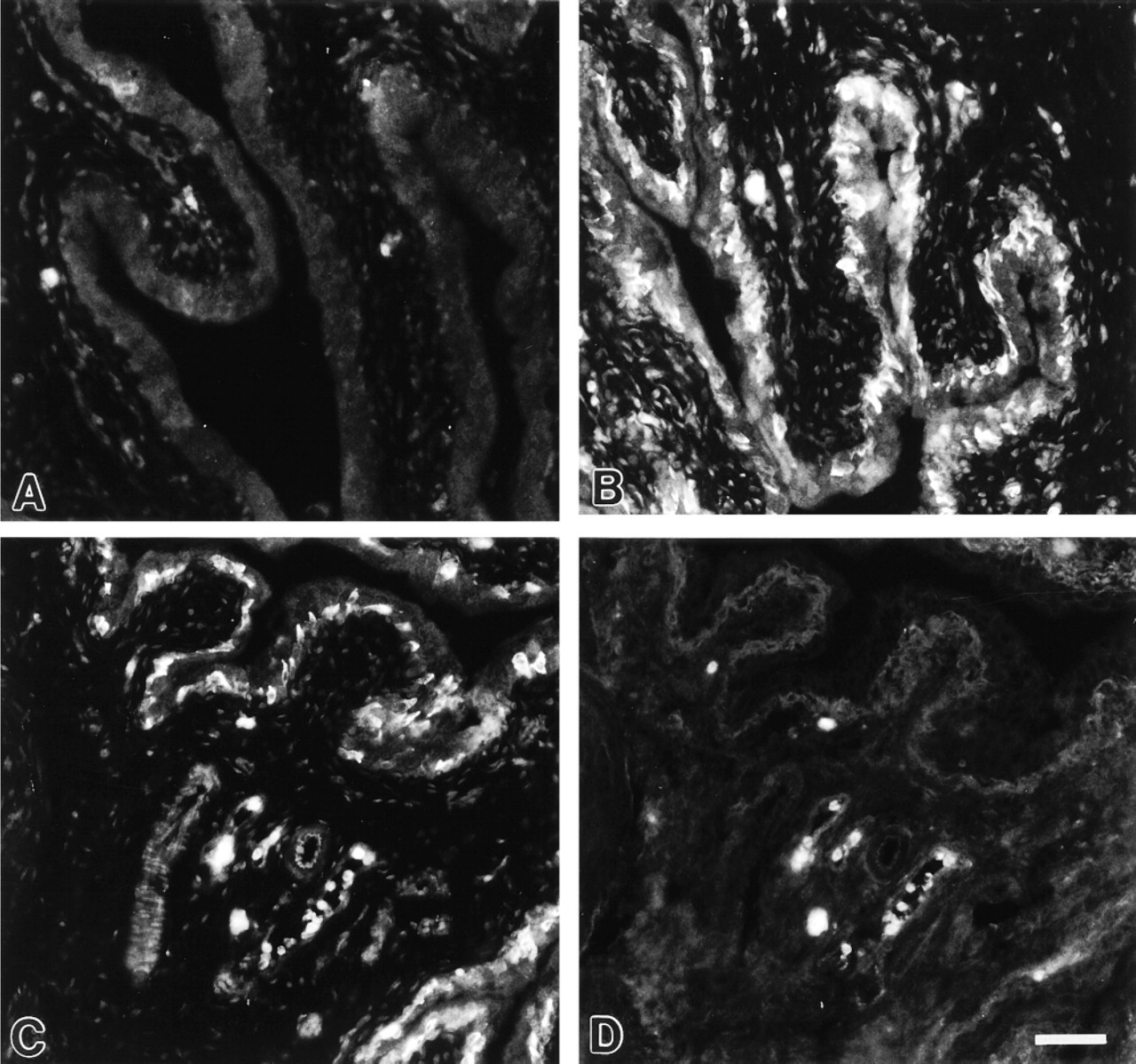
iNOS. Urothelial cells in control rats (given saline injection) were devoid of iNOS immunoreactivity (Figure 5A). In LPS-treated rats, the bladder urothelium displayed positive iNOS immunoreactivity (Figure 5B). The iNOS labeling showed a cytoplasmatic distribution and was found predominantly in cells located in the basal layer of the urothelium. Initial time course studies revealed that the immunoreactive response to iNOS was most prominent 6-9 hr after injection of LPS. There was no induction of eNOS or nNOS in urothelial cells in response to LPS. The monocyte/macrophage marker ED-1 (Dijkstra et al. 1985) was used to distinguish iNOS-positive urothelial cells from infiltrating macrophages. ED-1-positive cells, of which the majority expressed iNOS, were mainly associated with blood vessels. Double immunolabeling confirmed that iNOS-positive urothelial cells were negative for ED-1 (Figures 5C and 5D).
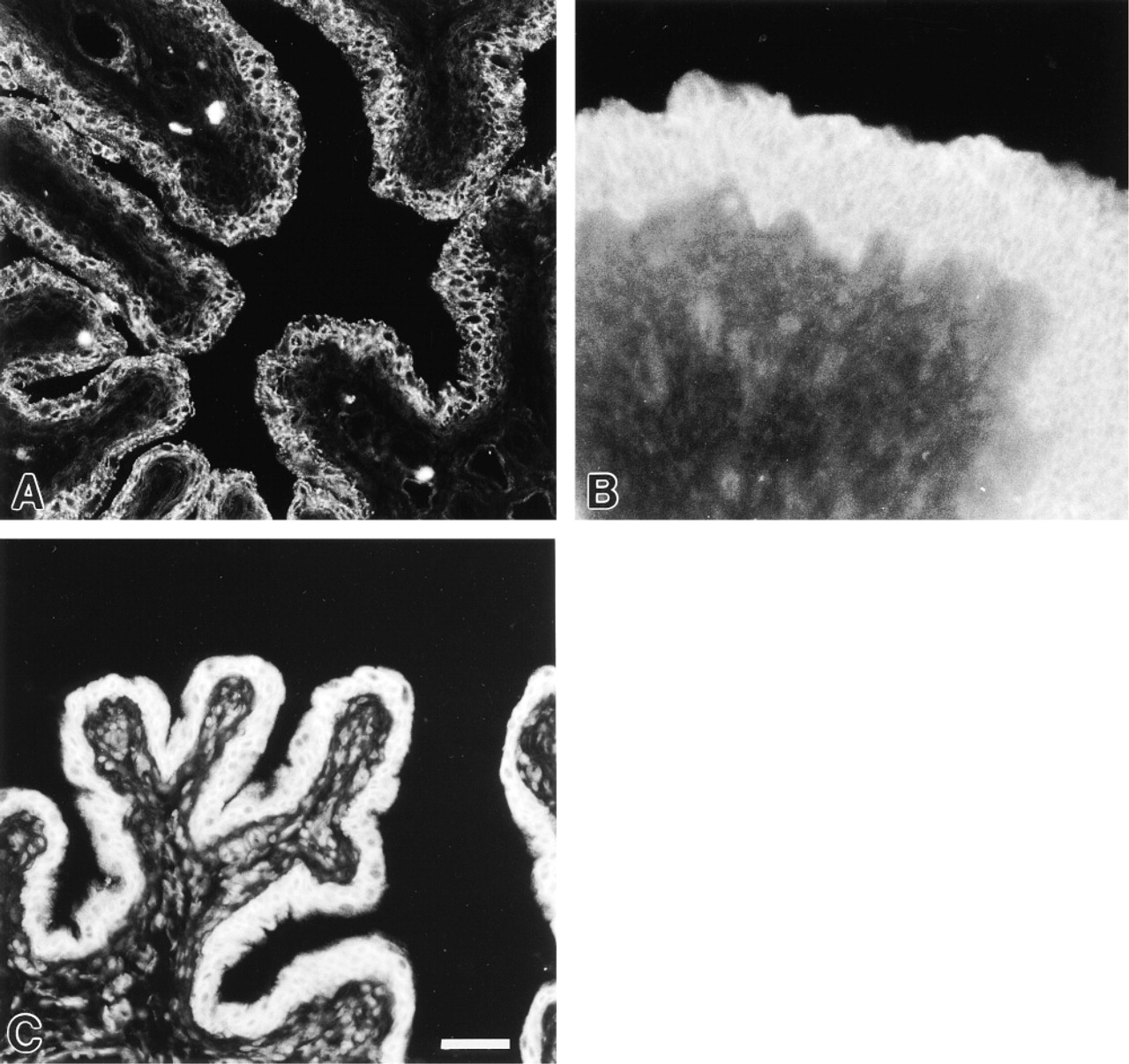
Cytokeratin 8.13 immunoreactivity of the rat (
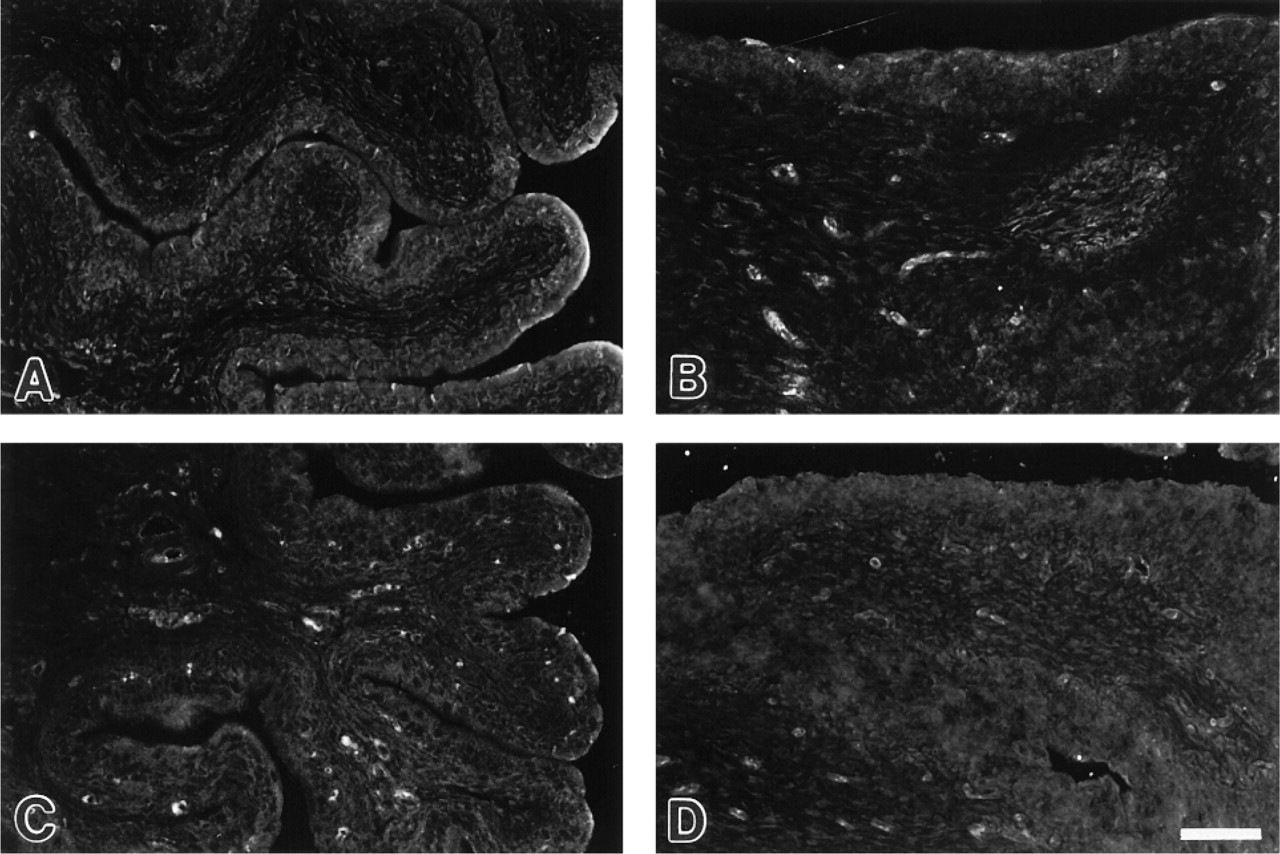
No immunoreactivity to nNOS was found in the rat (
Ultrastructural Demonstration of NADPH-d in Rat Urothelial Cells
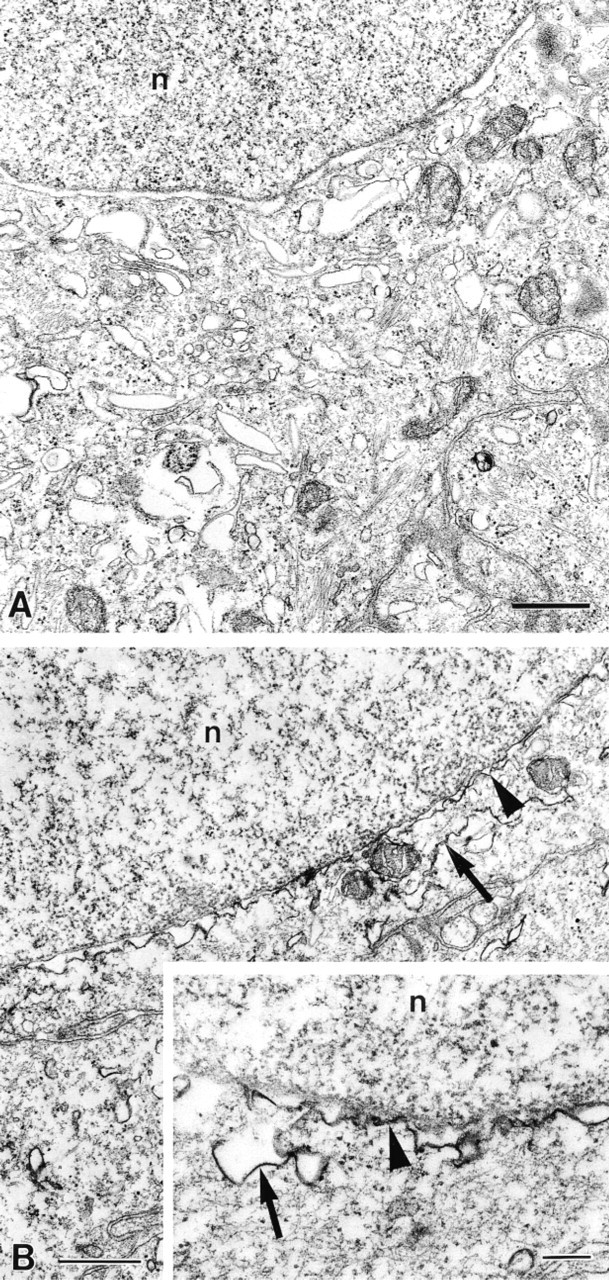
Ultrastructurally, the urothelial cells showed good tissue preservation of subcellular organelles, including the plasma membrane, nucleus, mitochondria, endoplasmic reticulum, and Golgi apparatus (Figure 6A). After incubation, the NADPH-d reaction product was found predominantly on membranes of the nuclear envelope and endoplasmic reticulum (Figure 6B). The reaction product also was found to be associated with the membranes of mitochondria. Omission of β-NADPH in control experiments resulted in unstained sections (Figure 6A).
Biochemical Detection of NOS in Pig Bladder
NOS activity was measured in specimens from the mucosa and the smooth muscle layer. Histological sectioning of the mucosal tissue used in the enzyme assay verified that it consisted of NADPH-d-positive urothelial cells. However, it was not possible to isolate the urothelial cell layer by dissection, and therefore the mucosal specimens also contained fractions of the suburothelial layer, i.e., nerves and vessels. The Ca2+ -dependent NOS activity in the cytosolic fraction of mucosal tissue averaged 5.6 ± 2.0 pmol/mg protein/min (n = 3), whereas the activity in the particulate fraction amounted to 1.6 ± 0.18 pmol/mg protein/min (Figure 7). The NOS activity was markedly reduced in the absence of Ca2+ (cytosolic 0.35 ± 0.05; particulate 0.57 ± 0.32 pmol/mg protein/min).
In samples representing the bladder smooth muscle, the cytosolic and the particulate Ca2+ -dependent NOS-activity amounted to 7.5 ± 1.5 and 2.4 ± 0.7 pmol/mg protein/min (n = 3), respectively (Figure 7). Ca2+ -independent NOS activity was negligible in both fractions.
Discussion
NADPH-d and NOS
The histochemical technique for NADPH-d activity has been widely used as a marker for the presence of NOS (Dawson et al. 1991; Hope et al. 1991). Despite the strong NADPH-d staining of the pig and rat urothelium, and although the various NOS antisera were proved to recognize endothelial and neuronal NOS, there was no indication of positive NOS labeling of urothelial cells. It might be argued that urothelial cells express a novel tissue-specific isoform of NOS that is not detectable with the antisera used in the present study. Indeed, a novel neuronal NOS isoform, different from the cerebellar NOS in containing an additional 34 amino acids, has been described in the urogenital tract (Magee et al. 1996) and in skeletal muscle (Silvagno et al. 1996). This novel isoform of NOS was, however, detectable with antiserum against cerebellar NOS (Silvagno et al. 1996). Nevertheless, it cannot be excluded that the different antisera used in the present study would fail to reveal a novel isoform of NOS in the urothelium. Biochemical detection of NOS activity using the conversion of radiolabeled L-arginine to L-citrulline (Bredt and Snyder 1989) detects all NOS activity in the tissue, and this method is therefore not limited to the specificity of antisera. In the pig bladder mucosa, a Ca2+ -dependent NOS activity was evident that was quantitatively comparable to the NOS activity found in the smooth muscle. However, it should be realized that the mucosal tissue samples, in addition to urothelial cells, also contained nerve fibers and vessels. As revealed by light microscopy, the relative amount of NADPH-d-stained tissue in mucosal specimens clearly exceeded the amount seen in muscle specimens. Therefore, if we assume that NADPH-d activity in urothelial cells is associated with NOS, higher rather than lower NOS activity would be expected in mucosal specimens. Moreover, NADPH-d activity associated with the NOS molecule is known to utilize only the β-isomer of NADPH (Beesley 1995). However, in a previous study we have found that the urothelial NADPH-d activity in pigs remained if β-NADPH was substituted with α-NADPH (Persson et al. 1993).
iNOS immunoreactivity in control (
Electron micrographs of rat urothelial cells labeled with the NADPH-d method. An unstained control section is shown in
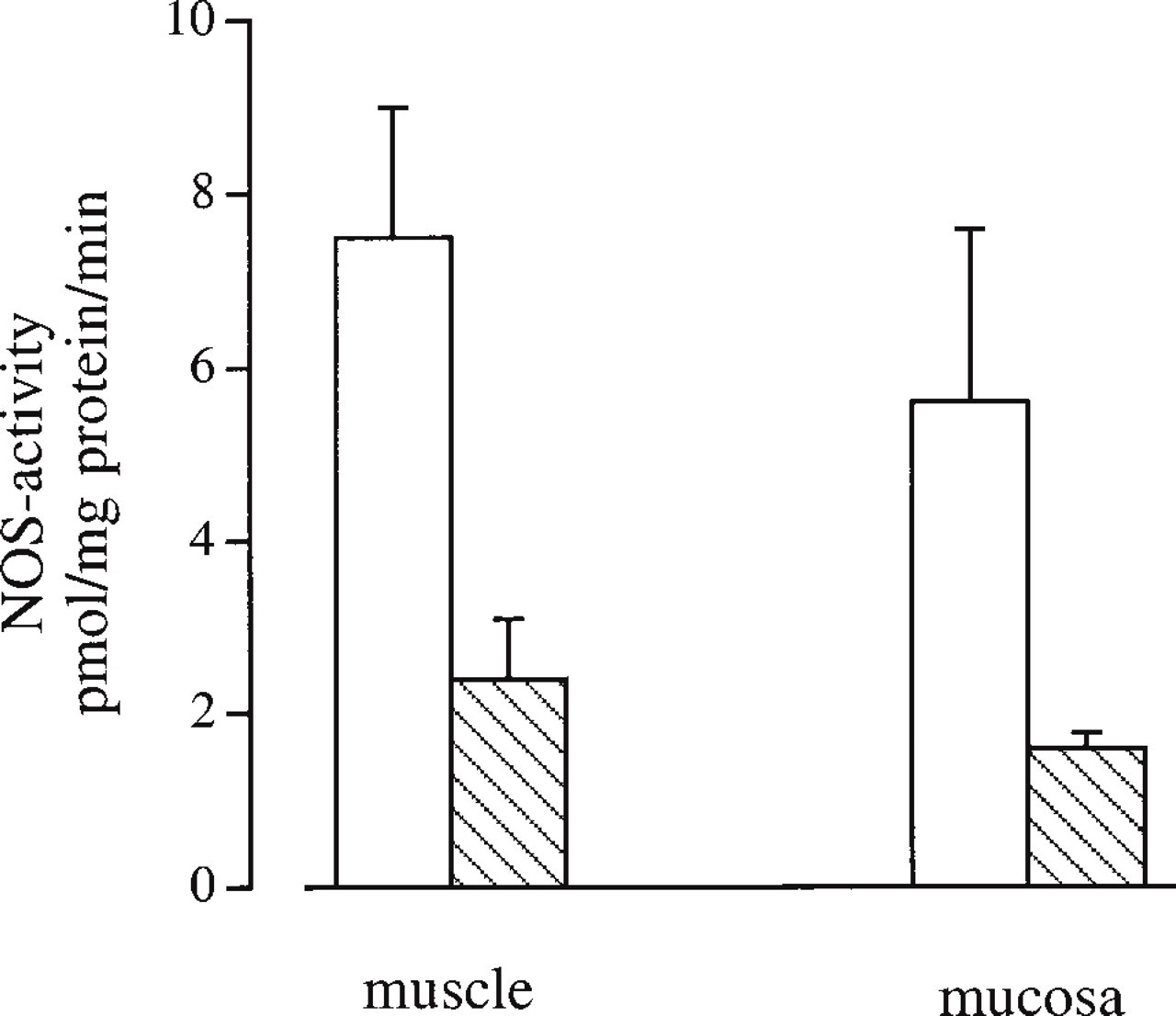
Enzyme assay of NOS activity in the pig bladder. Ca2+ -dependent L-citrulline production was measured in the cytosolic (open bars) and particulate (hatched bars) fractions of smooth muscle and mucosal tissue. Data are expressed as pmol I-citrulline/mg protein/min and are given as mean ± SEM (n = 3).
NADPH-d and Alkaline Phosphatase
Because the presence of NOS is unlikely to explain the urothelial NADPH-d staining, the involvement of other plausible enzymes that may account for the NADPH-d staining was further investigated. Alkaline phosphatase, which is expressed by urothelial cells (Page-Roberts 1972), may be responsible for the NADPH-d staining in certain non-neural cells (Grozdanovic and Gossrau 1995). The proposed mechanism is that alkaline phosphatase catalyzes the conversion of NADPH to NADH, causing NADH-d-derived formazan formation. Levamisole, an extraintestinal alkaline phosphatase inhibitor, was used to examine this possibility in rat urothelial cells. In sections preincubated with levamisole, the NADPH-d staining of the urothelium was clearly weaker, whereas the endothelial and neuronal staining was more or less unaffected. This suggests that the NADPH-d staining in urothelial cells is different from the endothelial and neuronal NADPH-d staining, and it also suggests that part of the urothelial NADPH-d staining may represent alkaline phosphatase activity.
NADPH-d and Cytochrome P450 Reductase
NOS has a significant amino acid sequence homology to NADPH cytochrome P450 reductase (Bredt et al. 1991) and, like NOS, this enzyme is a flavoprotein and is NADPH-dependent (Blottner et al. 1995).
NADPH-d staining has previously been found to coincide with cytochrome P450 reductase rather than NOS in various tissues, e.g., guinea pig epithelial cells (Young et al. 1997) and mouse olfactory epithelium (Kishimoto et al. 1993). In the present study we used an antiserum raised against NADPH cytochrome P450 reductase and found that this enzyme was uniformly and strongly expressed in the rat urothelium. Therefore, the urothelium has many similarities with other epithelial cells (Kishimoto et al. 1993; Young et al. 1997) in showing NADPH-d staining and NADPH cytochrome P450 reductase immunoreactivity but lacking expression of constitutive NOS. In general, cytochrome P450 reductase is not believed to possess NADPH-d activity in formaldehyde-fixed tissue, at least not in the brain (Matsumoto et al. 1993; Norris et al. 1994). There is a possibility that overlap between cytochrome P450 reductase immunoreactivity and NADPH-d activity in the rat urothelium is merely coincidental. In pigs, a discrepancy in cytochrome P450 reductase immunoreactivity between animals was noted, consistent with findings in guinea pig epithelial tissue (Young et al. 1997).
DPI is expected to inhibit flavoprotein-dependent enzymes (Stuehr et al. 1991), such as NOS and cytochrome P450 reductase, but it is not expected to affect flavoprotein-independent enzymes such as alkaline phosphatase. Preincubation with the flavoprotein inhibitor DPI totally abolished the endothelial and neuronal NADPH-d staining, and DPI also had pronounced effects on the urothelial NADPH-d staining. Therefore, the effect of DPI in combination with the previously discussed effect of levamisole favors the possibility that enzymes such as cytochrome P450 reductase and alkaline phosphatase contribute to the NADPH-d staining in the urothelium. In addition, various cytochrome P450 reductase-dependent enzymes such as, e.g., heme oxygenase, which catalyzes the cleavage of heme, may also be expressed by the urothelium. Our group has previously shown that the urothelium of the pig bladder displayed heme oxygenase-1 but not heme oxygenase-2 immunoreactivity (Werkström et al. 1997).
NADPH-d at the Ultrastructural Level
Nitroblue tetrazolium salt is considered to be unsuitable for ultrastructural localization of NADPH-d because it shows a tendency to dislocate. For ultrastructural NADPH-d studies, we used the BSPT salt, which yields an osmiophilic formazan on reduction and therefore reveals a more precise localization of the enzymatic reaction (Kalina et al. 1972). In rat urothelial cells, BSPT formazan was found to be associated with membranes of the nuclear envelope, endoplasmic reticulum, and mitochondria. This shows that the NADPH-d activity of the enzyme(s) responsible for the NADPH-d staining in urothelial cells is mainly associated with intracellular membranes. A similar subcellular distribution, related to portions of endocellular membranes, has been reported for both nNOS (Wolf 1997) and NADPH cytochrome P450 reductase (Phillips and Langdon 1962; Porter and Kasper 1985).
iNOS in the Urothelium
The inducible NOS isoform, iNOS, is Ca2+ -independent and is induced by bacterial endotoxins and cytokines in a number of cell types, including epithelial cells (Nathan and Xie 1994). Some epithelial cells, such as human airway endothelium (Guo et al. 1995) and mouse ileal mucosa (Hoffman et al. 1997), appear to express iNOS constitutively. However, in normal rats no immunoreactivity to iNOS was noted in the bladder urothelium. It has recently been reported that LPS administration causes upregulation of mRNA for iNOS in the rat bladder (Olsson et al. 1998). Furthermore, Cook and coworkers (1994) described iNOS immunoreactivity in rat urothelium after LPS-induced endotoxic shock, findings that were confirmed in the present study. The principal cells expressing iNOS in the bladder after LPS stimulation were urothelial cells and cells present in blood vessels. To exclude that iNOS labeling of the urothelium was due to infiltrating macrophages, some sections were double labeled with iNOS and ED-1, a marker for monocytes and macrophages (Dijkstra et al. 1985). These experiments verified that iNOS-positive urothelial cells were clearly separate from ED-1-positive cells. It has previously been shown that infected urine exhibits NOS activity (Smith et al. 1994,1996), with the primary source being excreted neutrophils (Wheeler et al. 1997). In addition, as shown in the present study, urothelial cells may represent an additional source of NO production during bladder infection. Induction of NOS is proposed to have cytotoxic properties against invading pathogens or tumor cells (Nathan and Xie 1994), but it may have a potential cost of damage to the host bladder by interference of NO with urothelial cell differentiation (Elgavish et al. 1996; Souza-Fiho et al. 1997). Indeed, urothelial cells may be targets for NO, as revealed by their immunoreactivity to cGMP (Smet et al. 1996). However, in our study, urothelial cells from both NO-stimulated bladders and LPS-treated rats were immunonegative for cGMP.
In conclusion, despite the strong NADPH-d staining of the rat and pig urothelium, no immunoreactivity to constitutively expressed NOS was detected in the urothelium. Activity of alkaline phosphatase and cytochrome P450 reductase may account for part of the NADPH-d reaction in urothelial cells. In LPS-treated rats, the urothelial cells express iNOS. The significance of NO production by urothelial cells in urinary tract infections remains to be established.
Footnotes
Acknowledgments
Supported by the Swedish Medical Research Council (grants 12601 and 11205), the Royal Physiographic Society, the Swedish Medical Society, the National Board of Health and Welfare, the Foundations of Crafoord, Magnus Berg-wall, Tage Blücher and Memorial Lars Hierta, and the Medical Faculty, University of Lund, Sweden.
We thank Dr J. de Vente (Section of Neuropsychology, Limburg University, Maastricht, The Netherlands) for supplying the cyclic GMP antibody.