
Abstract
We developed an improved method for the detection of double-strand DNA breaks in apoptotic cells at both the light (LM) and electron microscopic (EM) levels using a modification of the TdT (terminal deoxynucleotidyl transferase)-mediated dUTP nick end-labeling (TUNEL) technique. Cultured rat cerebellar granule cells were exposed to low potassium conditions to induce apoptosis. Twenty-four hr after treatment, one group of cells was fixed in situ with 4% paraformaldehyde and labeled for DNA fragmentation characteristic of apoptosis. Apoptotic cells were visualized with diaminobenzidine (DAB) and viewed by LM. The second group of cells was detached from the culture dish, pelleted, fixed with a 4% paraformaldehyde and 0.2% glutaraldehyde mixture, and embedded in LR White. For LM, the modified TUNEL technique was performed on 1.5-μm LR White sections and apoptotic cells were visualized using an enzymatic reaction to generate a blue precipitate. For EM, thin sections (94 nm) were processed and DNA fragmentation was identified using modified TUNEL with streptavidin-conjugated gold in conjunction with in-depth ultrastructural detail. Alternate sections of cells embedded in LR White can therefore be used for LM and EM TUNEL-based detection of apoptosis. The present findings suggest that the modified TUNEL technique on LR White semithin and consecutive thin sections has useful application for studying the fundamental mechanism of cell death.
A
A common hallmark of apoptosis is DNA fragmentation. Early in the apoptotic process there are double-stranded DNA breaks that generate large DNA fragments (50-sp300 KBP). Ultimately, the cell accumulates several million double-stranded DNA breaks that typically occur within the linker DNA regions between nucleosomes. Extraction of total DNA from an apoptotic cell population and analysis by agarose gel electrophoresis reveals a ladder pattern of approximately 180 BP and multimers thereof. Detection of the double-stranded DNA breaks in apoptotic nuclei can be readily achieved at the LM level on paraffin sections and cryosections using enzymatic detection of the free 3′ hydroxyl ends at the sites of the DNA breaks. A common method employed is to incorporate biotinylated nucleotides at the ends of the DNA using TdT, followed by binding to streptavidin-peroxidase conjugate and peroxidase detection using color development with diaminobenzidine tetrahydrochloride (DAB). Apoptotic cells are identified by a dark brown stain that is confined to the nucleus. Other similar methods employ alternative nucleotides and color development reagents but are all based upon terminal UTP nick-end labeling (TUNEL) using TdT (Gavrieli et al. 1992; Short et al. 1997).
LR White and LR Gold have been used by us and others as an embedding medium for histological stains such as hematoxylin and eosin (H&E), periodic acid-Schiff (PAS), trichrome stain, and the biochemical TUNEL stain (Migheli et al. 1995; Sanders and Wride 1996). However, successful detection of apoptosis by TUNEL- or ISEL-based methods on adjacent semithin and ultrathin LR White sections at both the LM and EM level has not been described. Because LR White medium has hydrophilic properties, we postulated that it might be adapted for in situ detection of the double-stranded DNA breaks associated with apoptosis in LM and EM studies. Thus, LR White could be used to detect double-strand breaks in DNA at both the LM and EM levels on serial thick and thin sections, respectively, from the same sample.
On the basis of our previous experience with LR White as an embedding medium for immunolabeling at the EM level (Goping et al. 1992,1996), we adapted a TUNEL-based technique for use on serial 1.5-μm and 90-nm sections of LR White-embedded cells and visualization by LM and EM, respectively. Labeling for apoptosis and visualization with EM corresponded at all times with detection of apoptosis at the LM level. We suggest that this new method will prove to be of great importance for future structural studies of apoptotic cell death.
Materials and Methods
Preparation of Cell Cultures
Cultures of rat cerebellar granule neurons were obtained from dissociated cerebella of 8-day-old Sprague-Dawley rats (Taconic Farms; Germantown, NY) as previously described (Gallo et al. 1982). Cells were plated in poly-L-lysine-coated two-well chamber slides for standard in situ detection of apoptosis using a TUNEL-based assay, or in poly-L-lysine-coated 12-well tissue culture plates for harvesting and embedding. At 18 hr after plating, cytosine arabinoside (10 μm) was added to the culture medium to inhibit the growth of non-neural cells. After incubation for 6–7 days, the cultures were either transferred into low potassium-containing medium (5 mM) to induce apoptosis (D'Mello et al. 1993), treated with 100 μm glutamate in the absence of glucose and magnesium to induce necrosis (Boje et al. 1993), or maintained in complete medium.
Preparation of LR White Sections
At 24 hr after induction of apoptosis and at 3 or 12 hr after induction of necrosis, cells were washed carefully with modified Locke's buffer (154 mM NaCl, 5.6 mM KCl, 2.3 mM CaCl2, 8.6 mM Hepes, pH 7.4) and harvested into 15-ml centrifuge tubes. Cells were centrifuged at 800 rpm for 3–5 min at room temperature (RT). Compact pellets were transferred directly into fixative. If necessary, loose pellets were resuspended in a small volume of culture medium, transferred into a microcentrifuge, and pelleted at 13,000 x g for 15 sec at RT. The pellet was carefully transferred to a small vial with a small wooden applicator. The cell pellets were fixed intact for 30 min at RT in 2.2% glutaraldehyde, pH 7.3, or in 4% paraformaldehyde, 0.2% glutaraldehyde, 2% sucrose, 0.1 M cacodylate buffer, pH 7.3, for EM or LM-EM labeling, respectively. After fixation, pellets were washed three or four times in 0.1 M cacodylate buffer. The first group was postfixed in 1% OsO4 in 0.1 M cacodylate buffer, 1% K3Fe(CN)6 to improve membrane contrast (Goping et al. 1995), and 1% sucrose. The second group was incubated in buffer containing only 1% sucrose in 0.1 M cacodylate buffer for labeling with the TUNEL-based assay. After 30 min, all samples were washed four times in cacodylate buffer. The fixed pellets were dehydrated in a graded ethanol alcohol series with 70% as the highest alcohol concentration, processed for LR White (Polysciences, Warrington, PA) embedding as described previously (Goping et al. 1996), and polymerized under vacuum at 40C.
Light Microscopy
Semithin (1.5 μm) sections from blocks with unosmicated LR White-embedded cells were cut with a Histoknife (DDK Delaware Diamond Knife) on a Reichert ultramicrotome. Extra care was taken to have a very clean knife and clean double-distilled water. Sections were picked up individually, transferred to a droplet of double-distilled water on a slide coated with poly-L-lysine, and dried on a hotplate for 20 min, then in a 40–50C oven for an additional 24 hr. One group of slides was overstained in Harris hematoxylin, briefly differentiated in 1% hydrochloric acid in 50% alcohol, then counterstained in eosin. A second set of slides was labeled using a modified TUNEL-based assay for detection of apoptosis. Semithin sections (1 μm) with osmicated LR White-embedded cells were stained with toluidine blue.
Electron Microscopy
Ultrathin sections were cut with a Diatome knife on a Reichert ultramicrotome at 94-nm thickness, collected on copper grids, and stained on an LKB ultra stainer with uranyl acetate and lead citrate (Leica, Deerfield, IL; standard solutions for LKB UltraStainer). Sections were examined and photographed in a CM100 Philips electron microscope. For apoptosis labeling, unosmicated LR White-embedded cells were cut and collected on nickel grids.
In Situ Apoptosis Staining for Attached Cells
Cells cultured in poly-L-lysine-coated two-well chamber slides were fixed in 4% formaldehyde in PBS for 10 min at RT, then immersed in 100% methanol for 5 min. The slides were washed once in PBS, and then the cells were covered with NeuroPore and left overnight at 4C. DNA labeling was then performed using the TACS (Trevigen Apoptotic Cell System) 2 TdT/DAB kit for apoptosis detection in situ. Details are found in manufacturers' instructions (Trevigen; Gaithersburg, MD).
In Situ Apoptosis Detection Modified for Cells Embedded in LR White
Modified TUNEL Assay for 1.5-μm LR White Sections on Slides. Sections were etched briefly (10 sec) in ethanol and rinsed in PBS. Sections were labeled for in situ detection of apoptosis using the TACS 2TdT/Blue Label kit (Trevigen) with slight modifications. Briefly, after three rinses in PBS, the sections were permeabilized in CytoPore for 30 min. Endogenous peroxide was quenched by immersion in 3% H2O2 in 40% methanol for 5 min, then rinsed in TdT labeling buffer. The sections were incubated in the TdT labeling mixture at 37C for 1 hr. The reaction was stopped and the sections were washed in water. The sections were covered with streptavidin-horseradish peroxidase diluted in Strep Blue Diluent according to the manufacturer's instructions and incubated at RT for 15 min. The sections were washed in water to remove unbound conjugate, then covered with TACS Blue Label. The development of the blue precipitate within nuclei of apoptotic cells was monitored under LM and was stopped after approximately 3 min by washing with water. Sections were counterstained with the Red Counterstain C (eosin-based) for 1 min.
TdT Assay for Thin Sections on Grids. For EM, a streptavidin-gold conjugate was used for direct visualization. The labeling reaction was performed as described above, except that permeabilization with CytoPore and quenching with H2O2 were eliminated. All washes of grids were performed in porcelain dishes, transporting the grids with a platinum loop. Drops of labeling reaction mix were placed on sheets of dental wax (Electron Microsopy Sciences; Fort Washington, PA), and the grids were placed section side down on the drops. Sections were incubated in a tightly closed humidified chamber overnight in an incubator at 4C with 2.5 μg/ml streptavidin-gold conjugate (EY Laboratories; San Mateo, CA) in PBS, 1% bovine serum albumin (BSA). When grids were placed from the last wash onto the TdT labeling mixture or streptavidin-gold conjugate, care was taken to remove excess liquid by touching the grid edges with lens paper. Sections were washed four times in PBS with 1% BSA, four times in PBS, three times in double-distilled water and stained in an LKB stainer (Leica) standard for 20 min with uranyl acetate and for 0.4 min with lead citrate.
Quantitation of apoptotic cells (Figure 3) in control, experimental apoptosis, and experimental necrosis cultures was carried out as follows. The total number of cells and apoptotic cells were counted in 4 L micrographs (+ OsO4), four-color TUNEL micrographs (- OsO4), and 6 E micrographs (+ OsO4) for control culture, necrosis culture, and apoptosis culture. The number of apoptotic cells was then expressed as a percentage of the total population. Every cell present in the micrograph was counted. The magnification of L micrographs was x 600 (304 cm2) of TUNEL micrographs x 590 (120 cm2), and the magnification of E micrographs was x 4750 (456 cm2).
Results
Cerebellar granule neurons in culture treated with low-potassium medium to induce apoptosis were labeled in situ for apoptosis using the TACS 2 TdT/DAB kit. Approximately 40% of the cells showed dark brown nuclear staining indicative of DNA fragmentation associated with apoptosis. The granule cells lost the network of neurites, the cell bodies were condensed, and the cell processes were shrunken (not shown). Therefore, apoptotic cells can be detected in situ in this system before embedding in LR White. Apoptosis has also been confirmed by others, using detection of DNA fragmentation by agarose electrophoresis of extracted total DNA (D'Mello et al. 1993; Sei et al. 1994).
For LM studies on LR White-embedded unosmicated cells, blocks were serial sectioned at 1.5 μm and alternately stained with H&E (Figures 1A-1C) or labeled with TACS 2 TdT/Blue Label (Figures 1D-1H). Control cells (Figure 1A) showed healthy granule cells and labeling for DNA fragmentation (Figure 1D) was negative, as indicated by the lack of a dark blue precipitate. Cells had a structured nucleus and clearly outlined membranes. In cells treated with low potassium to induce apoptosis and stained with H&E (Figure 1B), apoptotic cells were evident by the dark blue-stained and shrunken nucleus and clumping of chromatin. Adjacent sections labeled with the TACS 2 TdT/Blue Label kit (Figure 1E) showed a significant number of cells with blue nuclear precipitate indicative of apoptosis. The chromatin in apoptotic cells was clumped and was integrated with the nuclear membrane. Sections stained with peroxidase-conjugated streptavidin in the absence of the TdT enzyme showed no background blue staining (not shown), confirming that positive staining in the experimental sections was enzyme-mediated. The inset (Figure 1F) shows a high-power view of the typical morphology associated with apoptotic labeling, including condensed nuclei in shrunken cells. Therefore, embedding in LR White maintains apoptotic morphology when viewed by LM after detection of DNA fragmentation using TUNEL-based assays.
Necrotic cells, generated by exposure to glutamate for 3 hr and stained with H&E (Figure 1C), showed that many cells were lysed, with a reduction in cell numbers (see open spaces). Labeling of an adjacent section with the TACS 2 TdT/Blue Label (Figure 1G) also showed cell loss. In necrotic cells, cytoplasmic alterations precede the nuclear changes, in contrast to apoptosis, in which primary events occur in the nucleus (Anderson's Pathology 1996). The inset (Figure 1H) shows a high-power view of a labeled cell. There is particulate staining of chromatin on the nuclear membrane periphery of the remaining intact nuclei. The remaining cells are necrotic according to morphological criteria. Nevertheless, they do exhibit a degree of DNA fragmentation, as previously described (Eastman 1993). Therefore, although DNA fragmentation occurs in both apoptotic and necrotic cells, the high-quality LM images obtained with LR White sections allows the distribution of the label to be observed and distinguishes apoptosis from necrosis.
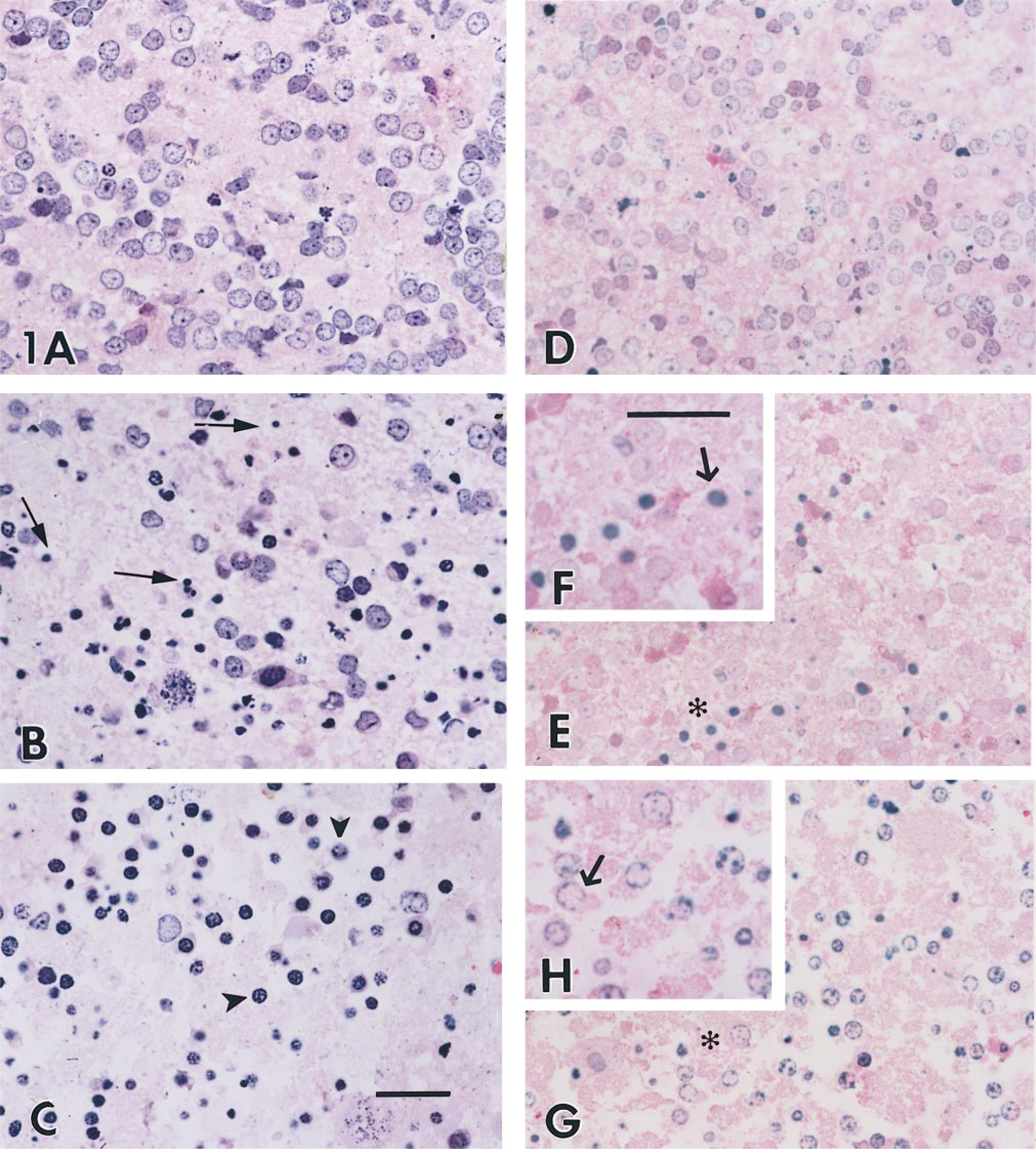
Plastic sections (1.5 μm) from granule cells embedded in LR White. (A-C) H&E staining; (D,E,G) stained for apoptosis. (A,D) Controls; (B,E,F) treated for apoptosis; (D,G,H) treated for necrosis (for 3 hr). (Insets) Enlargements of areas marked with asterisks. Thin arrows indicate typical apoptotic nuclei; arrowhead in C marks necrotic nuclei surrounded by lysed cytoplasm. Arrows in F and H indicate the typical apoptotic or necrotic cell, respectively. Bars = 25 μm.
The morphology was evaluated more thoroughly at the EM level (Figure 2). Control cells (Figure 2B) showed typical cerebellar granule cells with a large nucleus, limited cytoplasm, and a complete set of subcellular organelles. Cells treated with low potassium for 24 hr (Figure 2A) exhibited condensed and clumpy chromatin, cell shrinkage, and ruffled nuclear and cell membranes (Fesus et al. 1991). Many stages of apoptosis were identified by ultrastructure (Figure 2A). Disordered nuclear contours were prominent in two cells, with plasma membrane ruffling and blebbing in the same cells. Pyknotic nuclei of two cells indicated later stages of apoptosis. The cytoplasm of the cell at right was condensed; the organelles were seen as spindled remnants in a background of darkened ribosomes. Apoptotic nuclei are always noticeably electron-dense. The cell at upper right of Figure 2A showed apoptosis with secondary necrosis (Ankacrona et al. 1995; Anderson's Pathology 1996). The cell is swollen and the cytoplasm is disrupted, but the nucleus exhibits the electron-dense morphology associated with apoptosis. In contrast, a necrotic cell (Figure 2C) exhibited primary changes in the cytoplasm and disruption of the cell, nuclear, and plasma membranes. The cell had swollen and lysed cytoplasm and broken membranes (asterisk). Progressive membrane damage is a key event in necrosis. Chromatin condensation in the nuclear periphery corresponds to the labeling for DNA fragmentation observed previously (inset, Figure 1H).
We were interested in determining whether DNA fragmentation could be detected at the EM level from the same LR White blocks used to generate data for LM studies (Figure 1). For direct visualization of sites of DNA fragmentation, the TUNEL-based assay was performed using a streptavidin-gold conjugate. In control cells, no gold labeling was seen in either the cell nuclei or cytoplasm (data not shown), supporting the data presented above that the majority of cells in the control culture do not label with the TUNEL-based assay (not shown). In apoptotic cells (Figures 2D and 2E), gold particles were associated with the nucleus. The nucleus of the apoptotic cell in Figure 2E is shrunken and gold labeling is mainly seen in the nucleus, with a few grains in the cytosol (see arrows). Background labeling was almost nonexistent. In contrast to apoptotic cells, the necrotic cell shown in Figure 2F was lysed and the cell membranes were broken. A few gold particles were associated with the condensed chromatin and on the inside of the nuclear membrane. The deposition of gold particles in control, apoptotic, and necrotic cells is therefore in agreement with the respective blue labeling for LM (Figure 1). However, there is a distinct difference in labeling. The more necrosis proceeds, the more gold labeling decreases, in contrast to progressive apoptosis, in which gold labeling increases with time.
The results of the quantitative data for apoptosis are presented in Figure 3. In control samples, the number of apoptotic cells for both LM and EM was less than 5%, with numbers ranging from 2 to 4% of the total population counted. For necrosis samples, the percentage of apoptotic cells was also very low, ranging from 2 to 5%. EM exhibited the highest percentage of apoptotic cells, ranging from 25 to 31% of the total population. Although the data are not presented in the graph, necrotic cells were also counted in the experimental necrotic cultures. The percentage of necrotic cells in both the L and E micrographs ranged from 90 to 98% of the total cell count. As shown in the apoptotic data, the highest percentage was seen in electron micrographs.
Discussion
In this study, a technique to detect fragmented DNA in sequential sections for LM and EM from the same LR White block is described. The method allows the detection of fragmented DNA in 1.5-μm LR White sections and, simultaneously, a biochemical detection for DNA fragmentation in 90-nm sections visualized with streptavidin-gold conjugate. For end-labeling on semithin and ultrathin sections, we found it necessary to omit secondary fixation with OsO4 and/or membrane enhancement of the cells with potassium ferricyanide. DNA end-labeling on osmicated cells (different pellet, same culture) was 50% less efficient than labeling on unosmicated cells (not shown). However, for the ultrastructural observations alone and quantitative data (Figure 3), we employed OsO4 postfixation. DNA end-labeling of cells requires careful preparation of the cell pellet. Solvent exposure was kept at a minimum by employing short fixation, dehydration, and embedding times. The temperature for polymerization was maintained at 40C (in a vacuum oven). Treatment of sections with proteinase K was omitted because it exposes nonapoptotic cells, resulting in false-positive labeling.
Previous investigators have reported DNA end-labeling in apoptotic nuclei in tissue or cells at the LM level, using cryosections or paraffin-embedded sections (Wijsman et al. 1993). Others have described in situ labeling for apoptosis at EM using araldite, an epoxy matrix with hydrophobic properties (Migheli et al. 1995). We selected LR White for this methodology because it allows semithin and ultrathin sectioning and has hydrophilic properties, making it suitable for aqueous buffers used in the TUNEL. Using semithin and ultrathin sections of LR White, we were able to demonstrate labeling for DNA fragmentation at the LM and EM level, respectively. The massive number of DNA breaks that occur during apoptosis have made this biochemical event a common hallmark for apoptosis when other evidence may be difficult to obtain. Our results also show that DNA fragmentation can occur to some degree during necrosis, which may make interpretation of TUNEL on paraffin or cryosections at the LM level open to interpretation. The use of TUNEL on LR White sections allows assessment of both the distribution and the quantity of DNA fragmentation. The combination of morphological and biochemical data on the same section allows more detailed temporal analysis of the morphological and biochemical events that occur during apoptosis.
The detection of DNA fragmentation at the early stages of necrosis probably reflects the presence of free 3′ hydroxyl groups in DNA from nonspecific damage and the action of lysomal enzymes (Eastman 1993; Trevigen 1996).
Quantitative comparison of the number of apoptotic cells scored morphologically at the LM level using modified TUNEL, and morphologically at the EM level, gave similar results for control samples and necrotic samples, with the numbers of apoptotic cells being scored at below 5% regardless of the method of sample preparation. In apoptotic samples, the numbers of apoptotic cells determined using TUNEL was slightly less than that for morphological scoring at both the LM and EM levels. Because DNA fragmentation is known to be a relatively late event during apoptosis, it is likely that certain cells will exhibit sufficient morphological changes for the cell to be scored as apoptotic before DNA fragmentation. The highest percentage of apoptotic and necrotic cells was seen in the EM data. The most likely explanation for these data is the ease of identification of both the apoptotic and necrotic features at higher magnification.
One minor disadvantage of using the LR White matrix is that sections of embedded materials have to be kept relatively small. Therefore, more blocks must be cut.
The exact order of events in apoptosis is not known (Wijsman et al. 1993). Early changes have been reported to occur in the mitochondria by molecular or electrophysiological methods, which fail to be detected by morphological procedures (Enari et al. 1998; Sakahari et al. 1998). TUNEL or ISEL methods for morphological detection of apoptosis should be supplemented by additional criteria for detection of apoptosis by other methods (Yang et al. 1995).
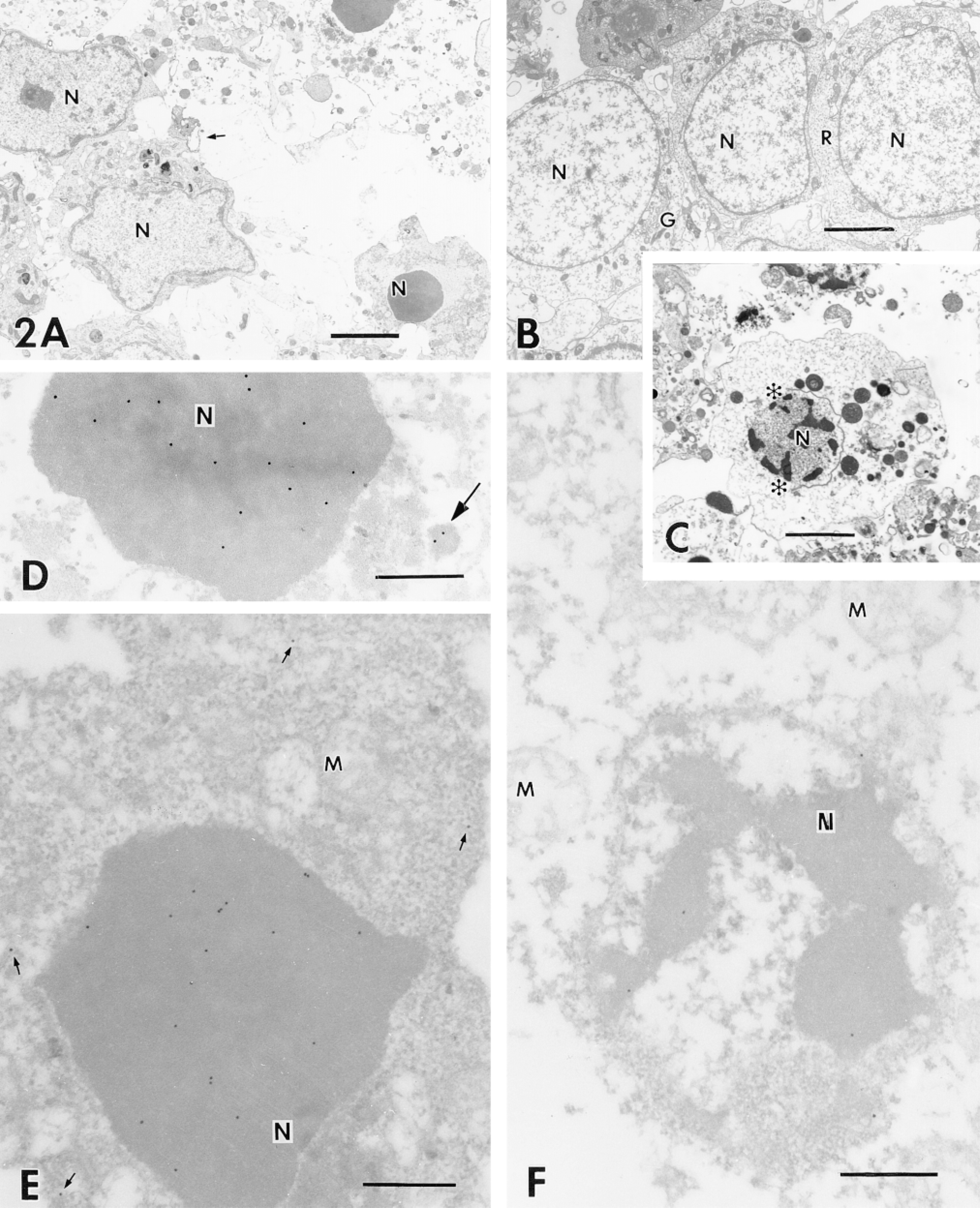
(A-C) Ultrathin sections of cells fixed in 2.2% glutaraldehyde, postfixed in OsO4 + K3Fe(CN)6, and embedded in LR White. (A) Cells treated in situ for apoptosis for 24 hr. Small arrow indicates blebs; (B) control; (C) cells treated for necrosis for 12 hr. Asterisks indicate loss of nuclear membrane. (D-F) Ultrathin sections fixed in 4% paraformaldehyde and 0.2% glutaraldehyde. Postfixation with OsO4 was omitted. Sections were labeled for DNA fragmentation using the modified TUNEL technique. (D,E) Apoptotic cells. Arrow indicates labeled nuclear fragment in cytosol; (F) necrotic cells. Small arrows point to gold labeling in cytosol. Nu, nucleus; G, Golgi system; er, endoplasmic reticulum; M, mitochondria; R, free ribosomes. Bars = 3 μm.
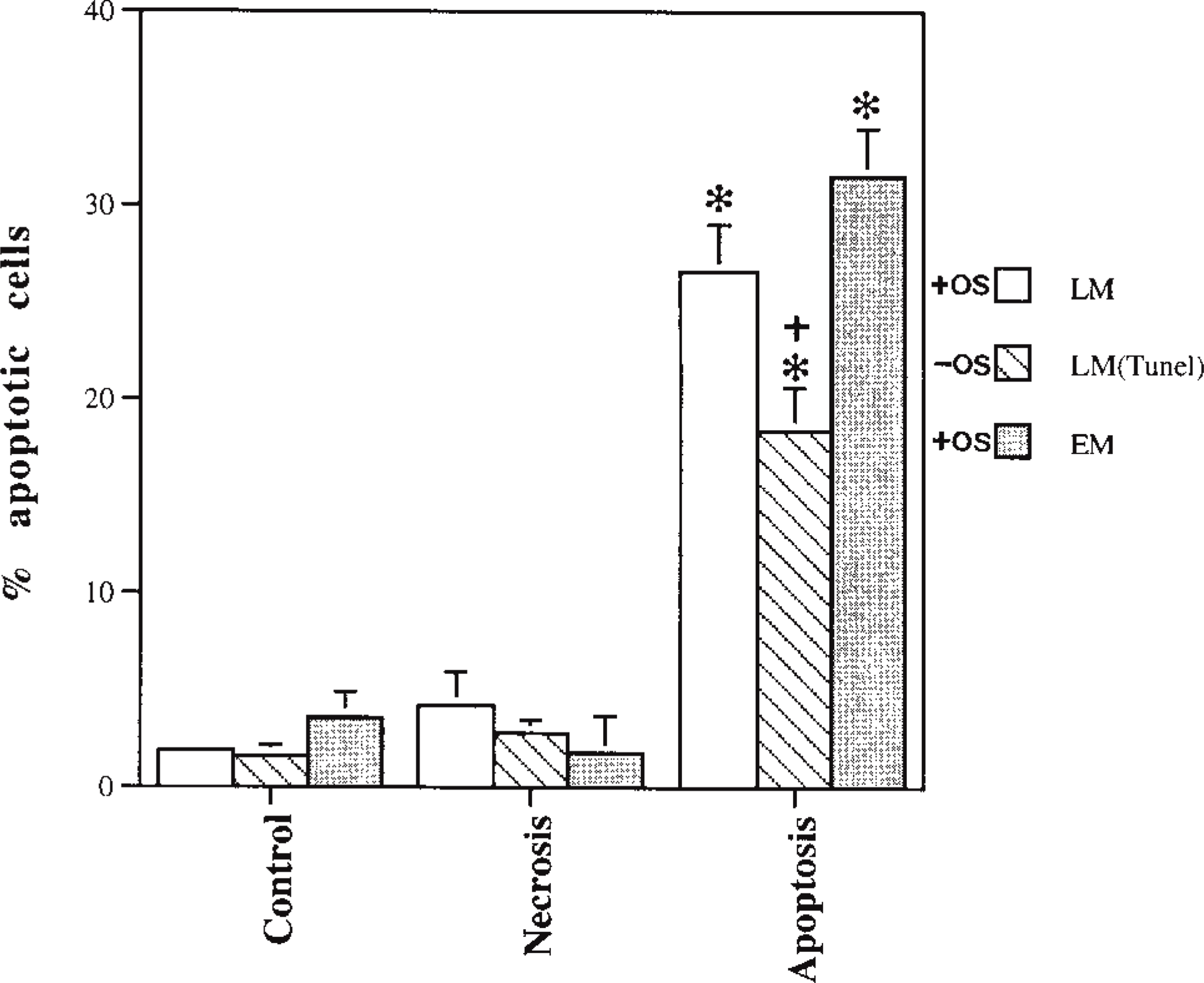
Percentage of apoptotic cells obtained by LM, LM-TUNEL, and EM in control, necrotic, and apoptotic samples. LM and EM sections from blocks with osmicated cells (+OS) are stained, respectively, with toluidine blue and uranyl acetate and lead citrate. LM-TUNEL sections are from unosmicated cells (-OS). The x-axis data range from 0 to 40 because of the low count in control and necrotic cultures. Data shown represent mean ± SEM of % positive (judged as apoptotic) cells in the examined field (n = 4–6) ∗p> 0.0001 compared to control and samples treated for necrosis; +p> 0.01 compared to measurement under EM. One-way ANOVA followed by Fisher's PLSD test.
It may be difficult to determine the type of cell death, as well as the degree and localization of DNA breaks, in such neurodegenerative diseases as Alzheimer's disease (Lucassen et al. 1997). Despite recent advances in techniques for detecting fragmented DNA, characterization of apoptosis and necrosis relies mainly on morphological (LM) and ultrastructural (EM) criteria (Ankacrona et al. 1995; Bonfoco et al. 1995). The classification of apoptosis has been historically determined by morphological criteria. In neurons, at least four types of cell death have been described (Martin and Johnson 1991), but to date there are limited data on the morphology at both the LM and EM levels. The biochemical detection of DNA fragmentation in association with morphological data enables the researcher to be specific in terms of cell death, and this is particularly important in the neuronal systems and when the classic morphology of apoptosis may not be obvious.
We suggest that the modified TUNEL-based assay used on LR White sections will prove to be an important tool in the study of mechanisms of apoptotic, necrotic, and other types of cell death in the CNS and other systems.
Footnotes
Acknowledgements
We thank Dr Juanita Anders for critical editorial assistance.