
Abstract
Endothelin-converting enzyme-1 (ECE-1) is the key enzyme of endothelin biosynthesis, catalyzing the final processing step. As shown by the targeted disruption of the ECE-1 gene, mature endothelins must be produced at specific sites for normal embryonic development. Therefore, it is important to know the exact pattern of ECE-1 gene expression. In this study we investigated the cellular distribution of ECE-1 in a variety of human tissues by in situ hybridization and immunohistochemistry. Widespread expression of the ECE-1 gene was noted, with a similar distribution pattern for mRNA and protein in normal human tissues, suggesting a major biological role for ECE-1. ECE-1 levels were particularly high in the cardiovascular, reproductive, and endocrine systems. There was strong and consistent labeling for ECE-1 in the vascular endothelial cells of all organs examined and in various nonvascular cells, especially some glandular cells. A large amount of ECE-1 protein and mRNA was detected in the Leydig cells of the testis and in the granulosa and theca cells of the ovary. In the adrenal gland, ECE-1 was detected in the cortex and medulla, with the strongest labeling in the zona glomerulosa. Therefore, ECE-1 may be involved in other systems, such as the regulation of hormone secretion, rather than exclusively generating ET-1 from its precursor. These results point out the potential side effects of ECE-1 inhibitors that are currently under development for treatment of cardiovascular diseases.
E
ETs are initially synthesized as large precursor polypeptides, called preproETs, which are cleaved at two pairs of basic amino acids, generating intermediate peptides, the bigETs. The bigETs are then cleaved by endothelin-converting enzyme (ECE), producing the mature ETs. ECE is a key enzyme in the biosynthesis of the ETs because the biological activities of big-ETs are negligible (Kashiwabara et al. 1989; Kimura et al. 1989). Two ECE genes have been cloned (ECE-1 and ECE-2). Their sequences are 59% identical (Emoto and Yanagisawa 1995), but only ECE-1 has been studied in detail (see Turner and Tanzawa 1997 for a recent review). ECE-1 is a novel membrane-bound metalloprotease (Ikegawa et al. 1990; McMahon et al. 1991; Opgenorth et al. 1992) structurally similar to neutral endopeptidase (NEP) (Malfroy et al. 1988) and Kell blood group protein (Lee et al. 1991). Its sequence is 39% identical to that of NEP and 32% identical to that of Kell protein. PEX, a phosphate-regulating gene identified as the candidate gene for X-linked hypophosphatemia, is a membrane-bound zinc metalloprotease and its sequence is 37% identical to that of ECE-1 (The HYP consortium 1995). ECE is inhibited by phosphoramidon but not by thiorphan, a specific inhibitor of NEP (Roques et al. 1993).
The distribution of ECE-1 mRNA in bovine and rat tissues has been investigated by Northern blot and RT/PCR analyses (Ikura et al. 1994; Xu et al. 1994; Shimada et al. 1995). There are particularly high levels of ECE-1 mRNA in the lung, adrenal gland, ovary, testis, and heart. In situ hybridization confirmed that ECE-1 mRNA is found in various bovine tissues, with the largest amounts in the endothelial cells and certain parenchymal cells (Xu et al. 1994). Takahashi et al. (1995) performed immunofluorescence studies with rat tissue sections and monoclonal antibodies (MAbs) raised against purified rat ECE. They detected ECE-1 protein in the endothelial cells of the aorta, lung, kidney, liver, and heart, and in certain endocrine cells, such as the chromaffin cells of the adrenal gland and the β-cells of the pancreatic islets.
The distribution of human ECE-1 has been analyzed by Northern blotting (Schmidt et al. 1994; Valdenaire et al. 1995) and RT/PCR (Rossi et al. 1995), showing that the ECE-1 gene is expressed in various tissues. However, these techniques detect mRNA in tissue homogenates, so they provide little information about the cellular distribution of ECE-1 in humans. Very recently, using an MAb raised against rat ECE, which recognizes rat and human ECE, the ECE-1 protein was detected in the smooth muscle cells of specimens from two patients with coronary atherosclerosis (Minamino et al. 1997) and in the epithelial cells of human epididymis (Peri et al. 1997). Pupilli et al. (1997) described the cellular distribution of ECE-1 mRNA and its protein in human kidney. ECE-1 mRNA and protein were detected in the vascular endothelium and tubule epithelial cells in the cortex and medulla of human kidney.
Targeted disruption of ECE-1 showed that it is the activating protease for both bigET-1 and bigET-3 in vivo (Yanagisawa et al. 1998). Surprisingly, large amounts of mature ET-1 peptide are found in ECE-1 −/− embryos, which are not produced at the locations crucial for normal embryonic development. This indicates that a detailed knowledge of ECE-1 tissue and cellular distribution is required, because mature ETs must be produced at specific sites for normal embryonic development. The aim of this study was to investigate the distribution of ECE-1 mRNA and protein in various human tissues, using techniques that facilitate the precise determination of cellular distribution. We used a specific antiserum directed against human ECE-1 (Korth et al. 1997), which does not crossreact with ECE-2 and NEP, for immunocytochemical studies to detect the ECE-1 protein. ECE-1 mRNA was detected in the same tissue specimens by in situ hybridization.
Materials and Methods
Human Tissues
Normal human tissues were obtained at surgery from tumor-free or disease-free resection margins from patients (n = 39) undergoing biopsy or resections for various reasons (e.g., localized tumor resection) or less than 24 hr after death from cadavers (n = 7) at the Institute of Pathology, Justus-Liebig-University (Giessen, Germany) or the Hôpital Tenon (Paris, France). The ages of the patients undergoing biopsy/resection ranged from 1 week to 80 years; 20 were male and 19 were female. The age of the deceased individuals ranged from 1 week to 49 years; five were male and two were female. We examined only tissue areas that appeared macroscopically and microscopically completely normal after routine histological staining. In postmortem tissues, autolytic tissue damage was excluded by hematoxylin-eosin-stained sections. In addition, protein integrity was checked by performing CD31 (1:10 in paraffin sections and 1:50 in frozen sections, clone JC/70A; DAKO, Hamburg, Germany) and vimentin (1:50 in paraffin sections and 1:100 in frozen sections, clone V9; DAKO) immunolabeling. Only positive stained specimens were used in the study. Tissues were either snap-frozen in liquid nitrogen, embedded in OCT Compound (Miles; Elkhart, IN), and stored at −80C until use or fixed in 4.5% formalin, pH 7.0, or 4% paraformaldehyde in PBS for at least 24 hr, routinely processed, and embedded in paraffin.
Immunohistochemistry (IHC) with CHO Cells
A stable CHO cell line that overproduces human ECE-1a (Korth et al. 1997) was used as a positive control to check the specificity of the 473-17-A ECE-1 polyclonal antiserum in paraffin-embedded specimens. Stably transfected CHO/ECE-1a and untransfected CHO cells were grown separately to confluence, scraped, mixed together (50:50), and centrifuged (10 min at 1000 × g). The pellet was fixed by incubation with 4% paraformaldehyde for 24 hr, dehydrated, and embedded in paraffin. Sections (5 μm) were cut and mounted on silanated slides. The paraffin was removed and the CHO sections were rehydrated and incubated for 20 min with 3% normal goat serum. They were then incubated with 473-17-A antiserum (1:1000) or preimmune serum (1:1000) for 90 min in a humid chamber. Antigen-antibody complexes were detected with the Vectastain ABC Elite kit (Vector Laboratories; Burlingame, CA) with diaminobenzidine as the substrate, as previously described (Sibony et al. 1995). The color reaction was stopped by rinsing sections in 50 mM Tris-HCl, pH 7.6.
Immunohistochemistry with Human Tissue Specimens
Cryostat or microtome sections (5 μm) of the specimens were mounted on slides, air-dried, frozen, and stored at −20C until required. Hematoxylin-eosin staining was used to exclude autolytic tissue damage in postmortem tissues. ECE-1 was detected (Bohle et al. 1997) with the highly sensitive alkaline phosphatase (APAAP) technique, using a modified version of the method of Cordell et al. (1984). Frozen sections were fixed in acetone for 10 min at room temperature (RT). Paraffin was removed from sections by incubation for 10 min in xylol, followed by 10 min in acetone and 10 min in 50% acetone/50% Tris-buffered saline (TBS), pH 7.4. The paraffin-embedded sections used for the detection of ECE-1 protein were boiled three times for 5 min each at 800 W in 0.01 M citrate buffer, pH 6.0, in a microwave. Frozen and paraffin-embedded sections were incubated for 30 min with the ECE-1 antiserum 473-17-A (1:1000 dilution). There were then three subsequent detection steps, each lasting for 30 min. The first incubation was with mouse anti-rabbit immunoglobulin (DAKO) 1:250 in RPMI-1640 medium (Life Technologies; Paisley, UK) containing 12.5% inactivated bovine serum to inhibit nonspecific reactions. The second was with rabbit anti-mouse immunoglobulin (DAKO; rabbit-“link,” 1:40 in RPMI-1640 medium containing 12.5% inactivated bovine serum), and the final incubation was with the APAAP complex (1:50; DAKO). The “link” and APAAP incubations were each repeated for 10 min. The samples were thoroughly washed in TBS, pH 7.4, between the steps. Alkaline phosphatase was allowed to react with new fuchsin (100 μg/ml) and levamisole (400 μg/ml) for 20 min at RT. Control sections were incubated with preimmune serum (1:1000) and mouse anti-rabbit immunoglobulin (clone MR12/52, 1 μg/ml; DAKO) as primary antibodies. Sections were counterstained with hematoxylin and mounted in gelatin. To check whether the 473-17-A ECE-1 antiserum would crossreact with ECE-2, undiluted antiserum was incubated with either ECE-1 peptide (amino acids 473–489), against which the antiserum was prepared, or with the corresponding ECE-2 peptide (amino acids 502–518) for 16 hr at 4C under shaking. Final peptide concentrations used were 1 mg/ml and 100 μg/ml. These preincubated peptides (ECE-1 or ECE-2)/antisera were incubated with frozen sections of adrenal gland (1:1000 dilution) for 30 min and IHC was performed as described above. Human neutral endopeptidase (NEP) was detected in frozen sections, with a mouse anti-CD 10 MAb (1:50, clone SS2/36; DAKO), and the link and APAAP steps were perfomed as described above.
Summary of ECE-1 mRNA and protein expression in human tissues a
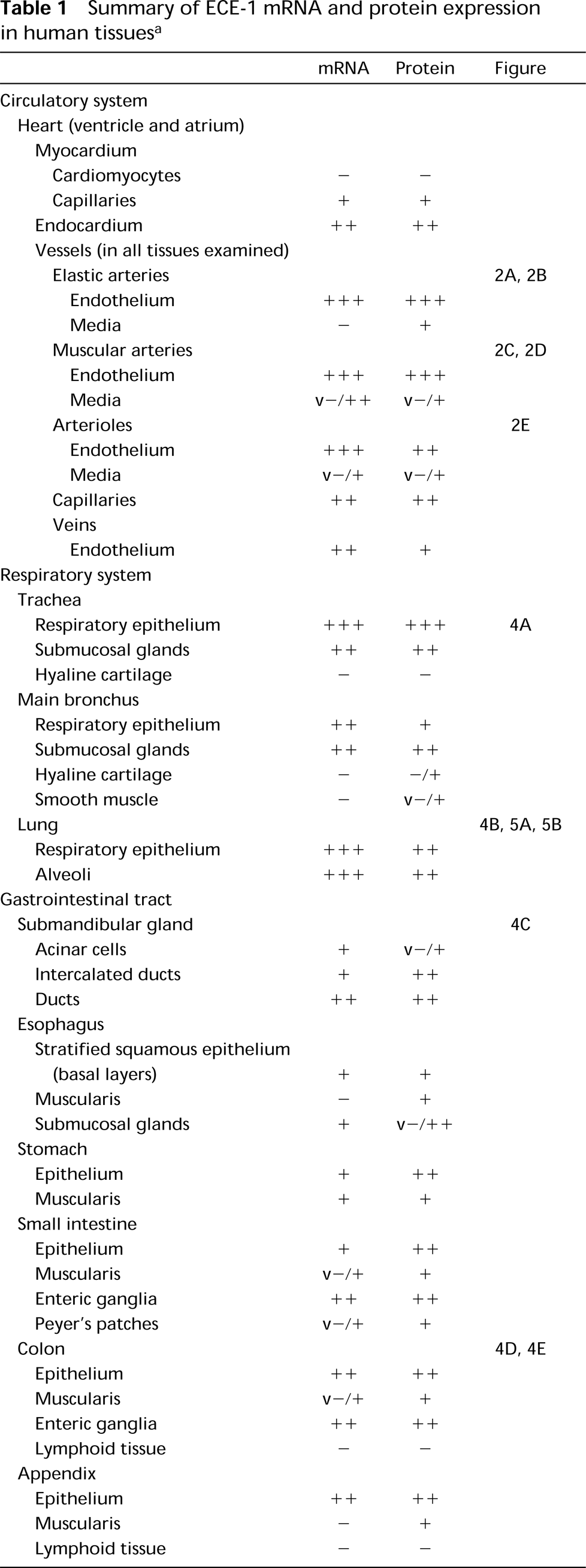
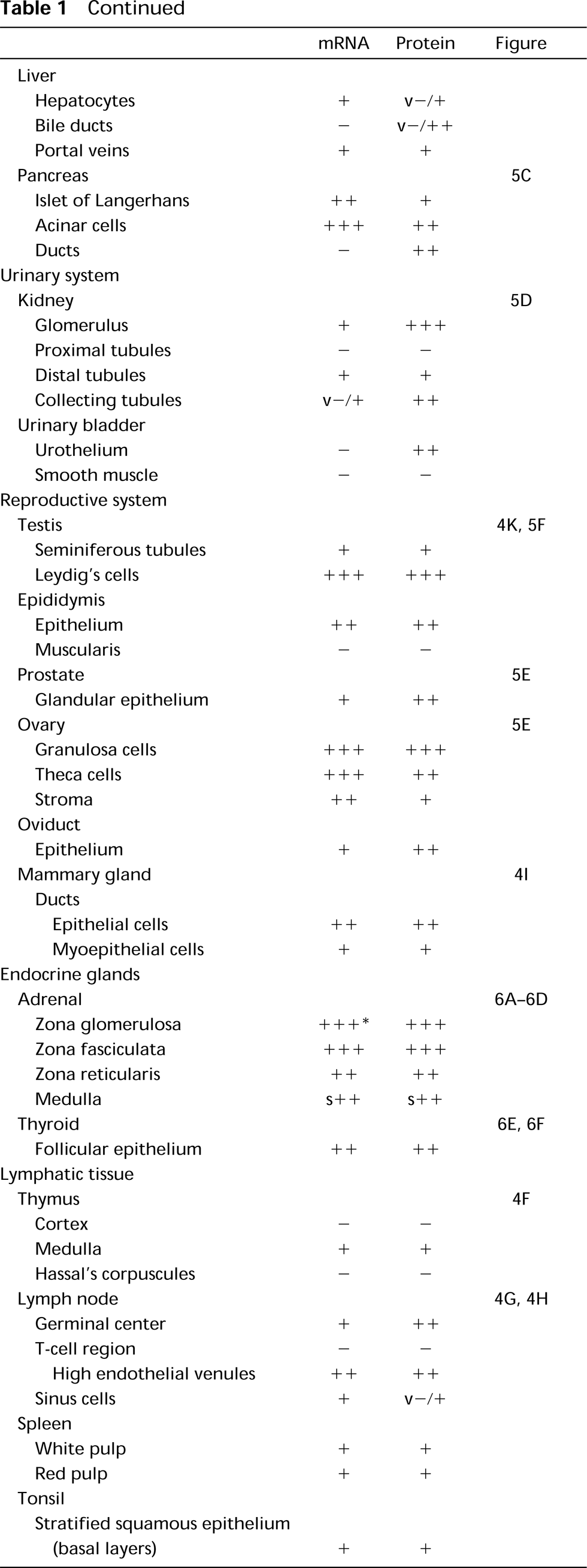
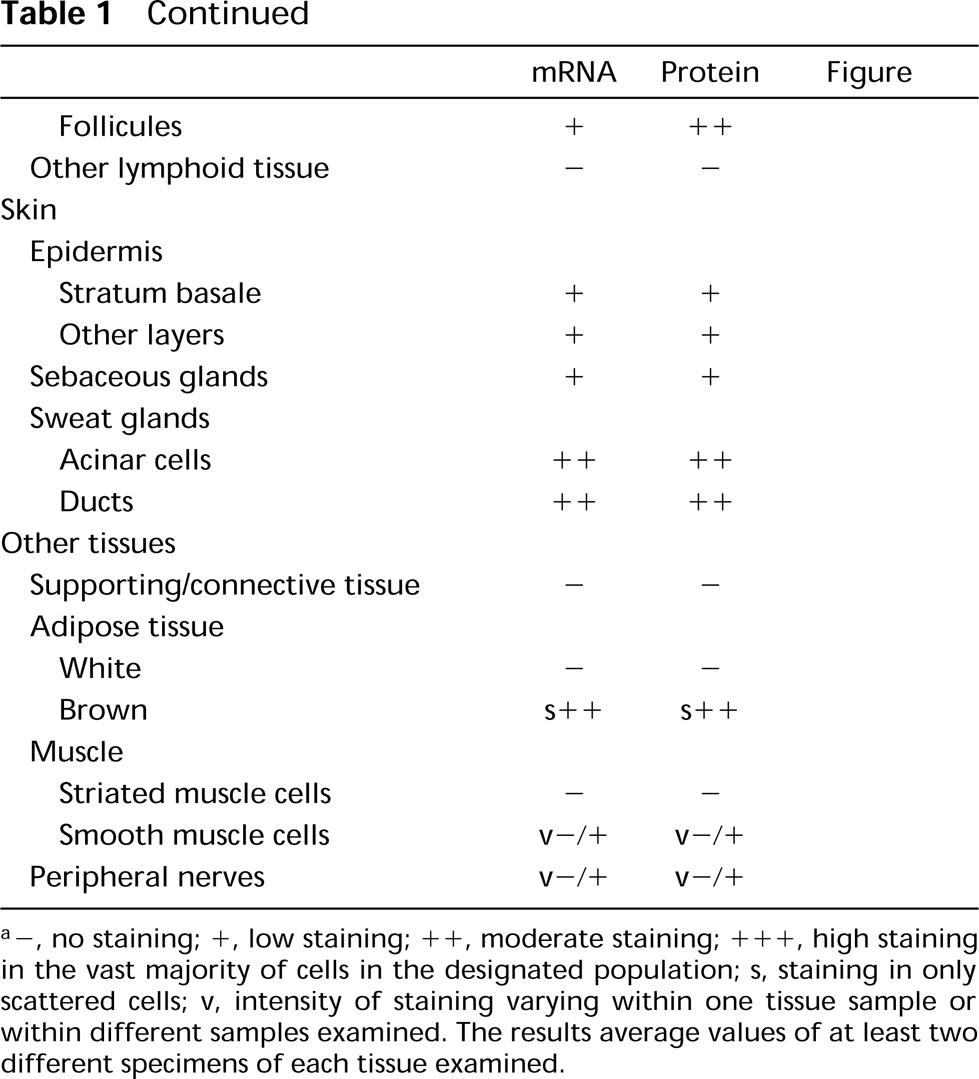
a −, no staining; +, low staining; + +, moderate staining; + + +, high staining in the vast majority of cells in the designated population; s, staining in only scattered cells; v, intensity of staining varying within one tissue sample or within different samples examined. The results average values of at least two different specimens of each tissue examined.
Preparation of Riboprobes
The human ECE-1a partial cDNA, corresponding to nucleotides 304–1666, was generated by RT/PCR using mRNA from primary cultures of human umbilical vein endothelial cells with the sense primer 5′-TCCATGGACCCCACGGTGGACCCC-3′ and the reverse primer 5′-AGGCGTTCACCATGGGCGGGGTCA-3′. It was subcloned into the pCRII vector using the TA cloning kit (Invitrogen; Leek, Netherlands). The sequence of the insert was checked using a Sequenase kit (US Biochemicals; Cleveland, OH). The recombinant plasmid was linearized by digestion with Hind III to obtain the anti-sense or Xba I to obtain the sense RNA. In vitro transcription and labeling with [35S]-UTP (Amersham; Les Ulis, France) were carried out together with T7 or SP6 RNA polymerase (Boehringer; Mannheim, Germany). Probes were precipitated with ammonium acetate and ethanol, dried by speed-vac centrifugation, and dissolved in TE-DTT (10 mM Tris, 1 mM EDTA, 20 mM DTT).
In Situ Hybridization (ISH)
We used an ISH protocol involving microwave pretreatment to enhance the hybridization signal, as described in detail elsewhere (Sibony et al. 1995). Paraffin-embedded sections (5–7 μm) were cut and two adjacent sections were mounted on each silane-coated slide. Deparaffinized sections were immersed in 0.01 M citric acid, pH 6.0, and heated in a microwave oven for 12 min. The sections were then incubated with proteinase K (2 μg/ml; Boehringer) and dehydrated. The two sections on each slide were hybridized by incubation overnight at 50C with the ECE-1 anti-sense and sense riboprobes (3–4 × 105 cpm per section). The slides were washed in a series of solutions, the stringency of each wash being higher than that of the last, and treated with RNase A (20 μg/ml; Sigma, Saint-Quentin, France). The sections were dehydrated and placed against Biomax film (Kodak; Rochester, NY) for 1–3 days. They were then dipped in NTB2 liquid emulsion (Kodak) and exposed for 1–3 weeks. Exposed slides were developed, fixed, and stained with toluidine blue.
IHC and ISH labeling was evaluated by different investigators (RMB, PK), each blind to the other's assessment. Results were classified on a 4-point scale, as shown in Table 1.
Results
Specificity of Anti-ECE-1 Antiserum and ECE-1 Riboprobe
The specificity of the polyclonal anti-ECE-1 antiserum 473-17-A (Korth et al. 1997) for human ECE-1 was checked in paraffin-embedded specimens from a stable CHO cell line that overproduces human ECE-1a, mixed with untransfected CHO cells (50:50), and routinely processed like a tissue sample.
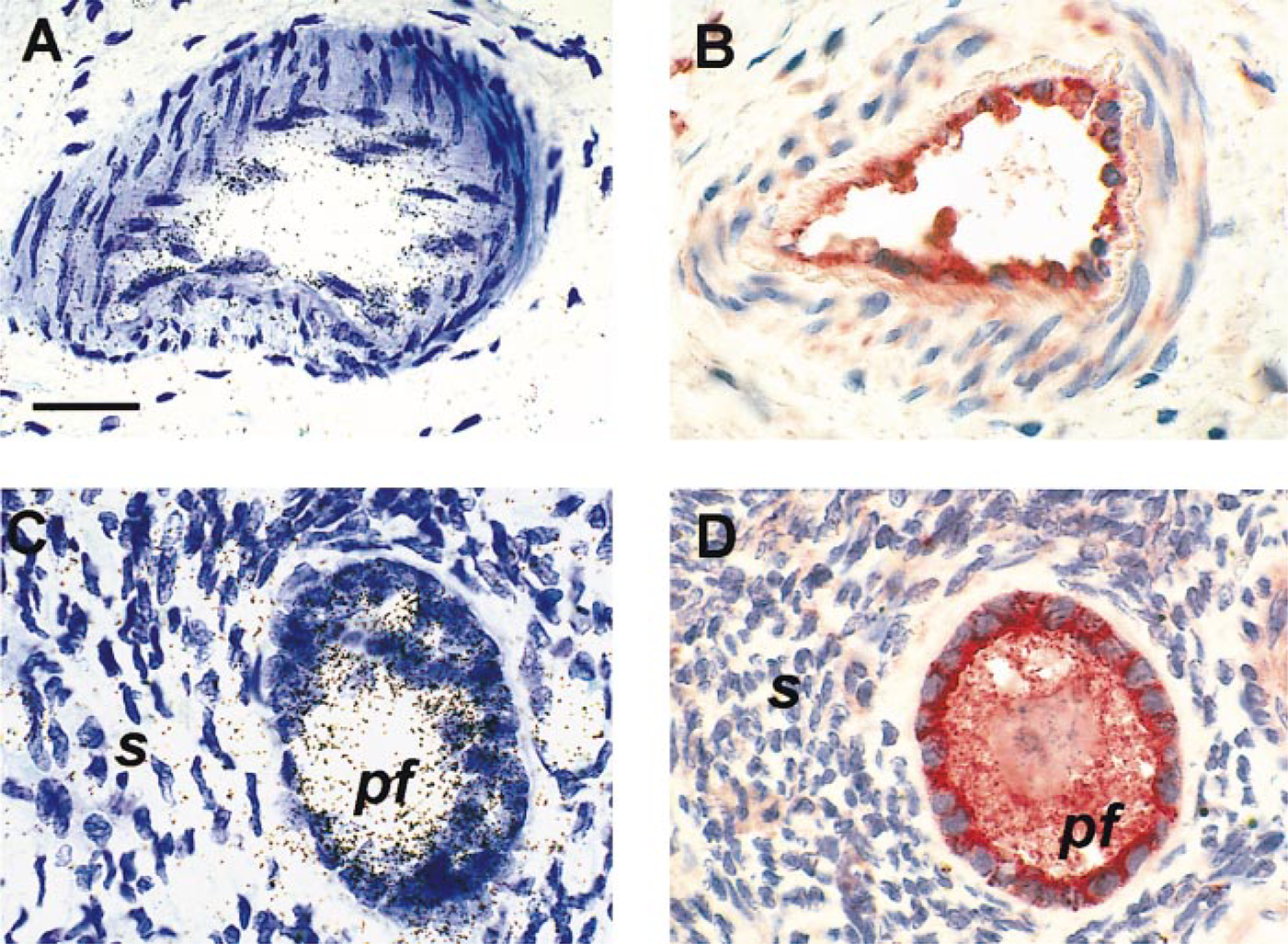
These sections were also used to ascertain the specificity of the ECE-1 RNA probe. The anti-sense ECE-1 probe (Figure 1A) and antiserum 473-17-A (Figure 1C) strongly labeled about half the cells, the proportion of cells producing ECE-1a. Therefore, antiserum 473-17-A and the anti-sense riboprobe for human ECE-1a were specific in paraffin-embedded specimens. Furthermore, the sense probe (Figure 1B) and preimmune serum (Figure 1D) did not react nonspecifically with transfected CHO cells overproducing ECE-1.
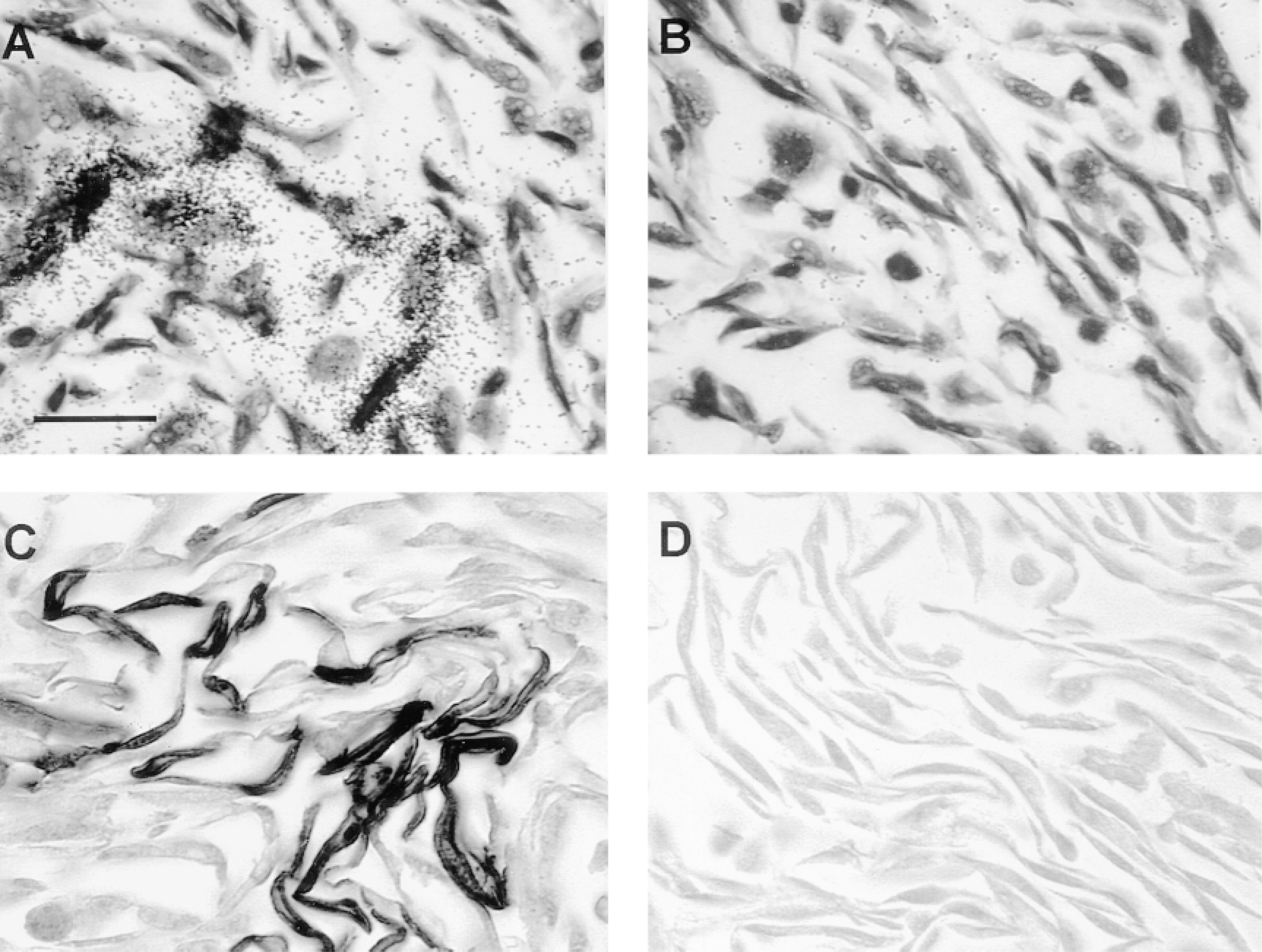
Specificity of the ECE-1 cRNA probe and anti-ECE-1 antiserum 473-17-A in paraffin-embedded specimens, tested with CHO cells. Transfected CHO/ECE-1 cells were mixed with untransfected CHO cells (50:50) and analyzed by in situ hybridization (A,B) and immunohistochemistry (C,D). Approximately half of the cells hybridized strongly with the anti-sense ECE-1 riboprobe (A); there was no signal with the sense probe (B). A similar proportion of CHO cells were immunolabeled with ECE-1 473-17-A antiserum in immunohistochemistry (C). No labeling was observed with preimmune serum (control) (D). (A,B) Cells were counterstained with toluidine blue. (C,D) No counterstaining performed for clearer immunostaining. Bars = 50 μm.
To exclude a crossreaction of anti-ECE-1 antiserum with ECE-2, we performed IHC with peptide-blocked antiserum on frozen sections of adrenal gland. When anti-ECE-1 antiserum was preincubated with the corresponding ECE-1 peptide, labeling was absent for a peptide concentration of 1 mg/ml and 100 μg/ml, whereas the same concentration of the corresponding ECE-2 peptide did not affect the labeling intensity (not shown). As a positive control, a serial section was labeled with untreated antiserum. This experiment demonstrated that the new anti-ECE-1 antiserum 473-17-A does not crossreact with ECE-2, despite the high homology between the two enzymes within the pep-tide region used for ECE-1 antiserum generation.
In a previous study, crossreaction of anti-ECE-1 antiserum with NEP was checked by immunoblotting of purified NEP (Korth et al. 1997). The antiserum 473-17-A did not recognize human NEP up to a concentration of 2 μg of pure protein. We also checked for crossreaction between ECE-1 and NEP by IHC with antiserum 473-17-A and anti-CD10 MAb, respectively, in serial frozen sections of all the tissues examined in this study. A different staining localization was observed with the antibodies, showing the differential distribution of the two human zinc metallopeptidases and the specificity of the two antisera (not shown). Therefore, anti-ECE-1 antiserum 473-17-A and the ECE-1 RNA probe were specific for ECE-1 only.
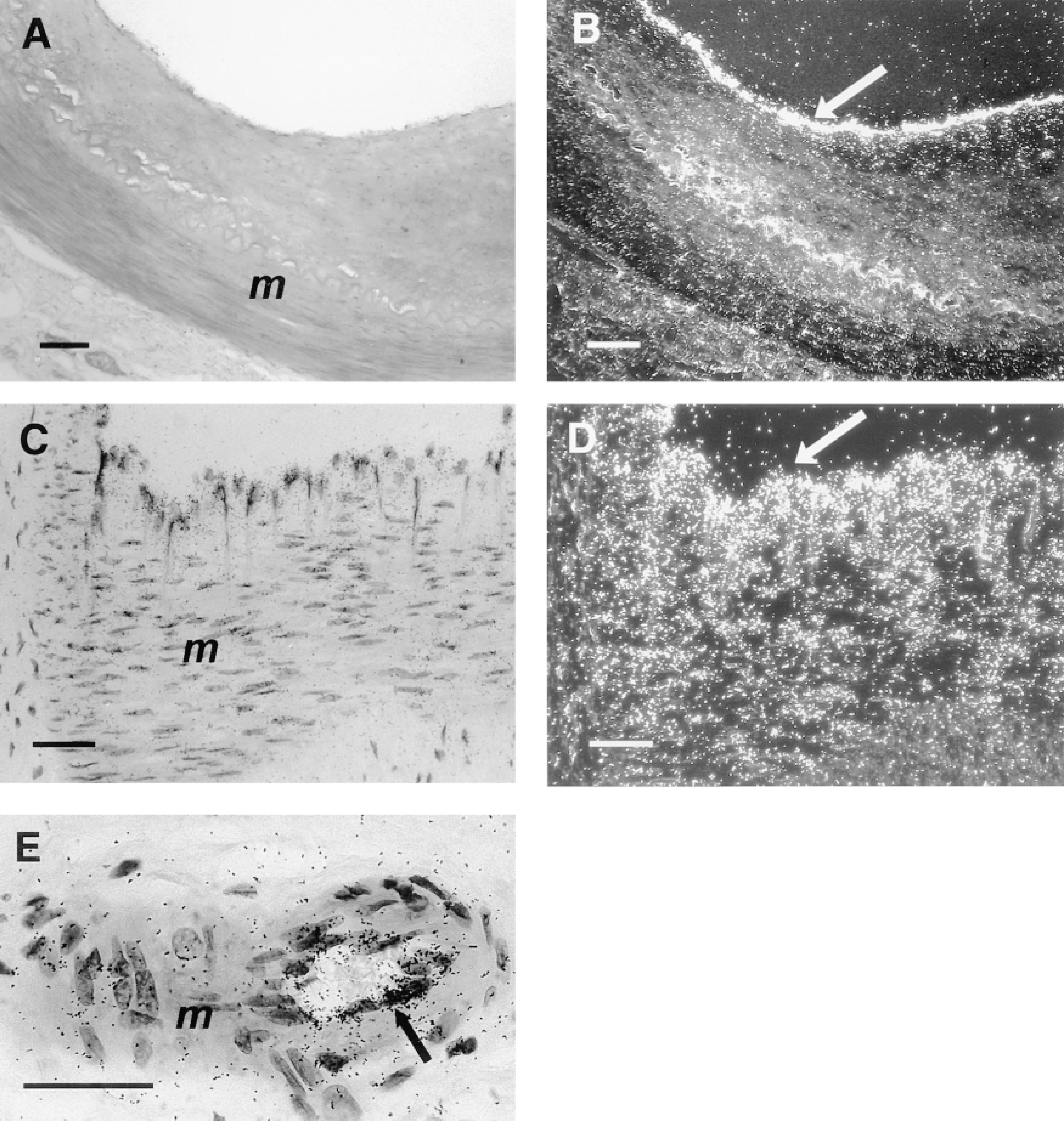
Human ECE-1 mRNA in vessels of the circulatory system. In situ hybridization with the anti-sense riboprobe was performed in serial sections of elastic artery (A,B), muscular artery (C,D), and arteriole (E). Strong signal was detected in the endothelium of all vessels (arrows). Media (m) of elastic artery (B) and arteriole (E) shows only unspecific background labeling, whereas the media of the muscular artery (D) shows focal, weak ECE-1 mRNA labeling. (A,C,E) Brightfield illumination; (B,D) darkfield illumination. Bars: A,B = 100 μm; C,E = 50 μm.
Synthesis of ECE-1 mRNA and Protein in Human Tissues
We compared the immunostaining pattern of freshly fixed cryostat sections with that of formalin-fixed, paraffin-embedded sections of the same material. The distribution and intensity of labeling were similar in each case (not shown). ISH and IHC were performed for at least two paraffin-embedded specimens of each tissue examined. We looked for the presence of ECE-1 mRNA and protein in the same cells, by performing ISH and IHC on serial sections for some tissues (Figures 3 and 6).
The distribution patterns of ECE-1 mRNA and protein in normal human tissues are summarized in Table 1. The average staining intensity values, as estimated by two independent investigators in two to seven tissue sections from different patients, are given. Figures 2 to 6 show photomicrographs of typical examples of ECE-1 mRNA or protein synthesis in all major organs. In almost all tissues examined, ECE-1 mRNA and ECE-1 protein had identical distributions. Their relative staining intensities were equivalent except for some epithelia (e.g., those of the bladder), in which there were differences between mRNA and protein levels. ECE-1 mRNA and protein were widely distributed in epithelial and nonepithelial tissues and in almost all major organs. However, neither ECE-1 mRNA nor protein was detected in certain nonvascular mesenchymal tissues, such as supporting/connective tissue, white adipose tissue, striated muscles, and hyaline cartilage.
Circulatory System
A high level of ECE-1 mRNA and protein was detected in almost all vascular endothelial cells, regardless of the organ of origin. There were only minor differences in the ECE-1 labeling intensities of endothelial cells from conducting (Figures 2A-2D) or resistance vessels (Figures 2E, 3A, and 3B). Moreover, ECE-1 labeling intensity was higher in the endothelial cells of microvascular arteries than in the venous endothelial cells. However, in some specialized endothelial cells from the glomeruli of kidney and the sinusoidal cells of the liver and spleen, the level of ECE-1 mRNA and protein synthesis was less predictable (Table 1). Moderate levels of ECE-1 mRNA and protein were also detected in the endocardium (Table 1). In contrast to the situation in the vascular endothelial cells, ECE-1 mRNA and protein levels in vascular smooth muscle cells differed greatly, from undetectable to moderate within the same vessel or within several vessels from the same specimen, whereas the myocardium produced no ECE-1 (Table 1).
Human ECE-1 mRNA and ECE-1 protein in vessels (A,B) and the reproductive system (C,D). In situ hybridization of ECE-1 with anti-sense riboprobe (A,C) and immunohistochemistry using anti-ECE-1 473-17-A antiserum (B,D) were performed on serial sections of normal small muscular artery (A,B) and normal ovary (C,D). In the vessel, ECE-1 mRNA (A) and ECE-1 protein (B) were restricted to the endothelial cells, with no labeling of smooth muscle cells. In the ovary, a large amount of ECE-1 mRNA (C) and ECE-1 protein (D) was detected in the granulosa cells of the primary follicle (pf), whereas the surrounding stroma (s) was only moderately labeled. Bar = 50 μm.
Respiratory System
ECE-1 mRNA and protein are synthesized throughout the respiratory system. The respiratory epithelium, from the trachea (Figure 4A) down to the bronchioli, was moderately to strongly labeled for ECE-1 mRNA and its protein. The smooth muscle cells of the tunica muscularis in the conducting airways were irregularly labeled for ECE-1 protein, whereas its mRNA was not detectable (Table 1). ECE-1 mRNA was absent from hyaline cartilage, whereas the seromucous glands of the trachea and main bronchus synthesized both mRNA and protein (Figure 4A). There was strong labeling for ECE-1 LmRNA in the alveoli of lung (Figure 4B) and moderate staining for ECE-1 protein (Figure 5A) in almost all cells. No staining was observed when preimmune serum was used (Figure 5B). We were unable to distinguish accurately between capillary endothelial cells and pneumocytes. However, ECE-1 was synthesized in almost all the cells of the alveolar region, so it is probable that, in addition to endothelial cells, at least one type of pneumocyte also synthesizes ECE-1.
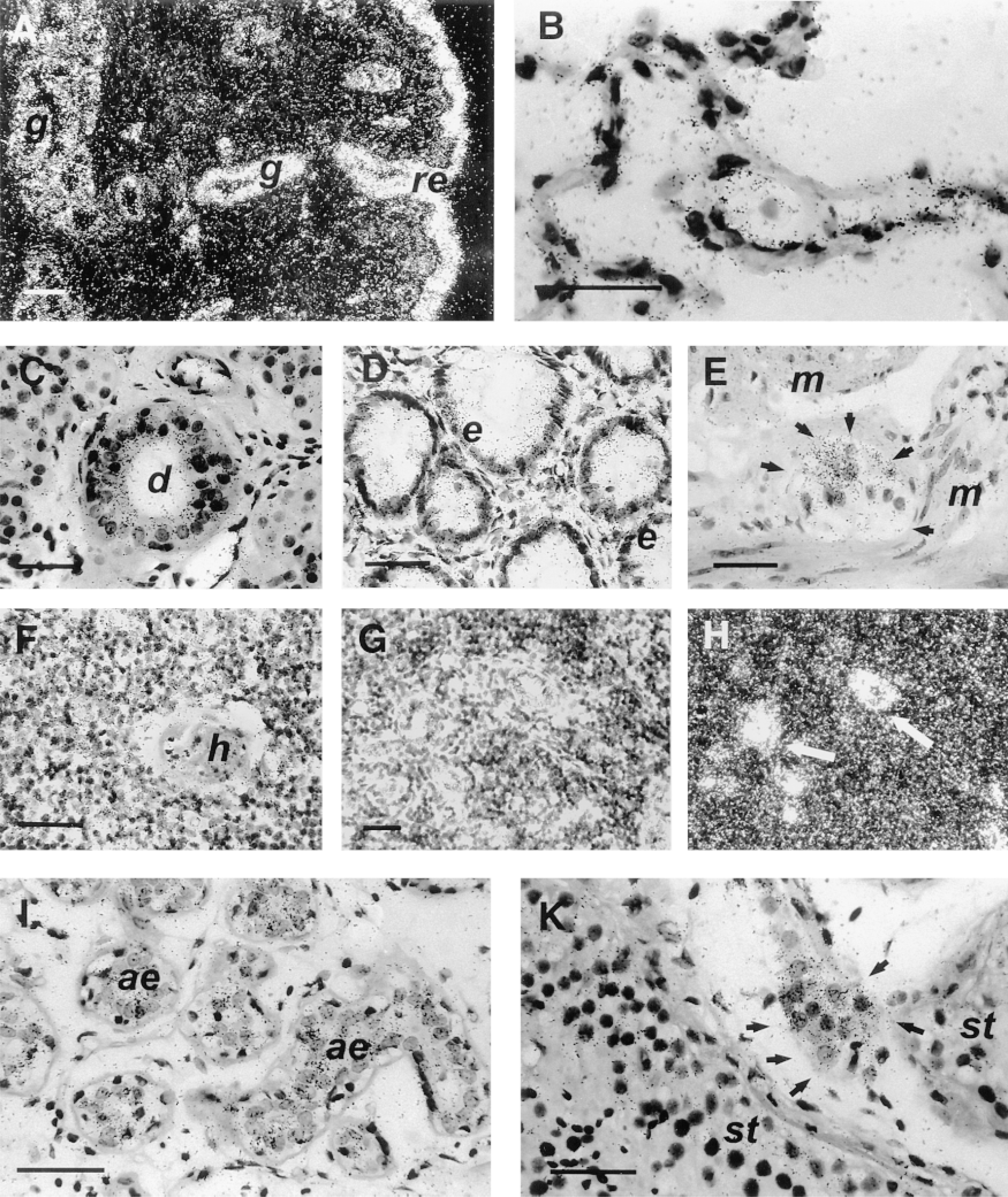
Human ECE-1 mRNA in the respiratory system (A,B), gastrointestinal tract (C-E), lymphatic tissue (F-H) and reproductive system (I,K), detected by in situ hybridization with the anti-sense probe. (A) At low magnification, intense labeling of the respiratory epithelium (re) and moderate labeling of the submucosal glands (g) of the trachea. Bar = 100 μm. (B) At high magnification, normal lung with high ECE-1 mRNA levels in the alveoli. (C) In the submandibular gland, moderate labeling of the epithelial cells of ducts (d), with acinar cells only weakly labeled. (D) In the colon, moderate levels of ECE-1 mRNA in epithelial cells (e) and scattered labeling in the lamina propria. (E) Enteric ganglion cells (Auerbach's plexus) in the large intestine are strongly labeled (arrows), whereas no signal is detected in the two layers of the muscularis (m). (F) Thymic medulla is weakly labeled and Hasall's corpuscle (h) is unlabeled. (G,H) Photomicrographs of serial sections of the paracortical zone of the lymph node showing ECE-1 mRNA in high endothelial venules (arrows). (I) In the mammary gland, moderate levels of ECE-1 mRNA are detected in acinus epithelium (ae). (K) Leydig's cells (arrows) in testis are more strongly labeled for ECE-1 mRNA than seminiferous tubules (st). (A,H) Darkfield illumination; (B,C-K) brightfield illumination. Bars = 50 μm.
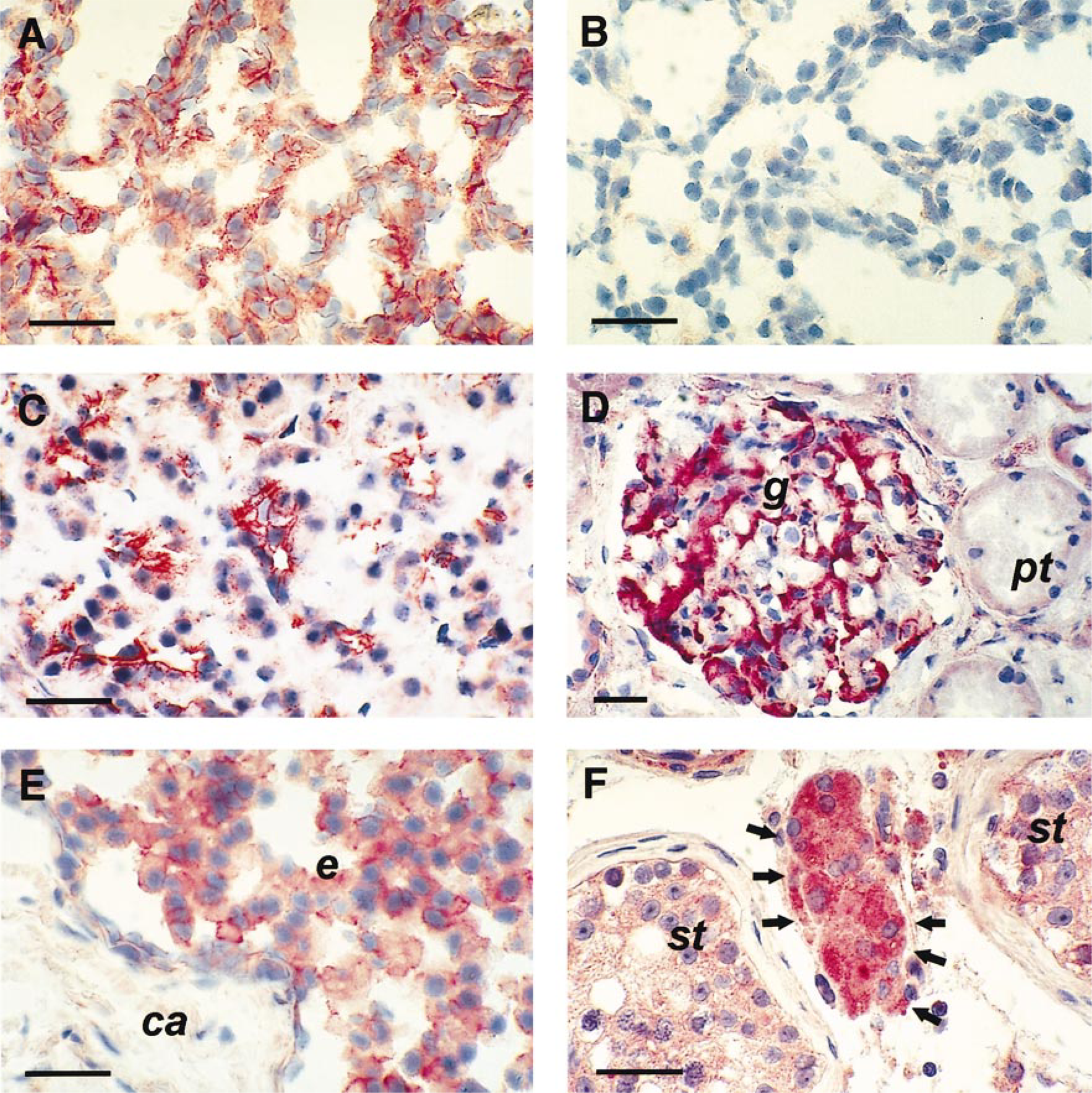
Human ECE-1 protein in the respiratory system (A,B), gastrointestinal tract (C), urinary tract (D), and reproductive system (E,F). Immunohistochemical analysis was performed with the anti-ECE antiserum 473-17-A. Normal lung with strong immunostaining in alveoli (A) and no specific immunostaining of the adjacent section when preimmune serum was used as a control (B). Moderate ECE-1 protein levels in the acinar cells of the exocrine pancreas (C). In the kidney, strong immunostaining is detected in the glomerulus (g), whereas the proximal tubules (pt) have only background levels of immunostaining (D). The glandular epithelium (e) of the prostate is moderately stained, whereas the capsule (ca) contains no ECE-1 protein (E). Leydig cells (arrows) in the testis are strongly stained for the ECE-1 protein, whereas seminiferous tubules (st) are only weakly stained (F). Bars = 50 μm.
Gastrointestinal Tract
ECE-1 was detected in the secretory and collecting parts of the submandibular gland. ECE-1 mRNA synthesis was moderate in intercalated and large duct epithelial cells but was low in acinar cells, whereas ECE-1 protein synthesis was variable in the acini and moderate in all ducts (Figure 4C). The squamous epithelium of the esophagus contained a low level of ECE-1 in its basal cell layers. The smooth muscle cells of the muscularis produced small quantities of ECE-1 protein, but no mRNA was detected. However, both mRNA and protein were detected in the submucosal glands (Table 1). The intensity of ECE-1 mRNA and protein labeling in the tunica muscularis was low (Table 1). There was low to moderate labeling of ECE-1 mRNA in the intestine (Figure 4D), with no consistent difference between the basal and luminal parts of the intestinal epithelium. Little or no ECE-1 was detected in the intestinal muscularis. Lymphoid tissue (known as Peyer's patches) in the small intestine was weakly labeled for ECE-1 mRNA and protein in a diffuse pattern, whereas ECE-1 was absent from the lymphoid aggregations of the large intestine (Table 1). The strongest labeling was detected in the ganglia of the mesenteric plexus (Figure 4E) and in the submucosal plexus. In the liver, little or no ECE-1 was present in hepatocytes. There was low labeling in the endothelial cells of the portal vein and no mRNA was detected in the bile ducts, whereas the intensity of ECE-1 protein labeling was absent to moderate in the various specimens (Table 1). The secretory acini of the exocrine pancreas were strongly labeled for ECE-1 mRNA and moderately stained for the protein (Figure 5C). The ECE-1 protein was also detected in the duct epithelium, but its mRNA was undetectable. In the endocrine pancreas, there was weak to moderate staining for ECE-1 (Table 1).
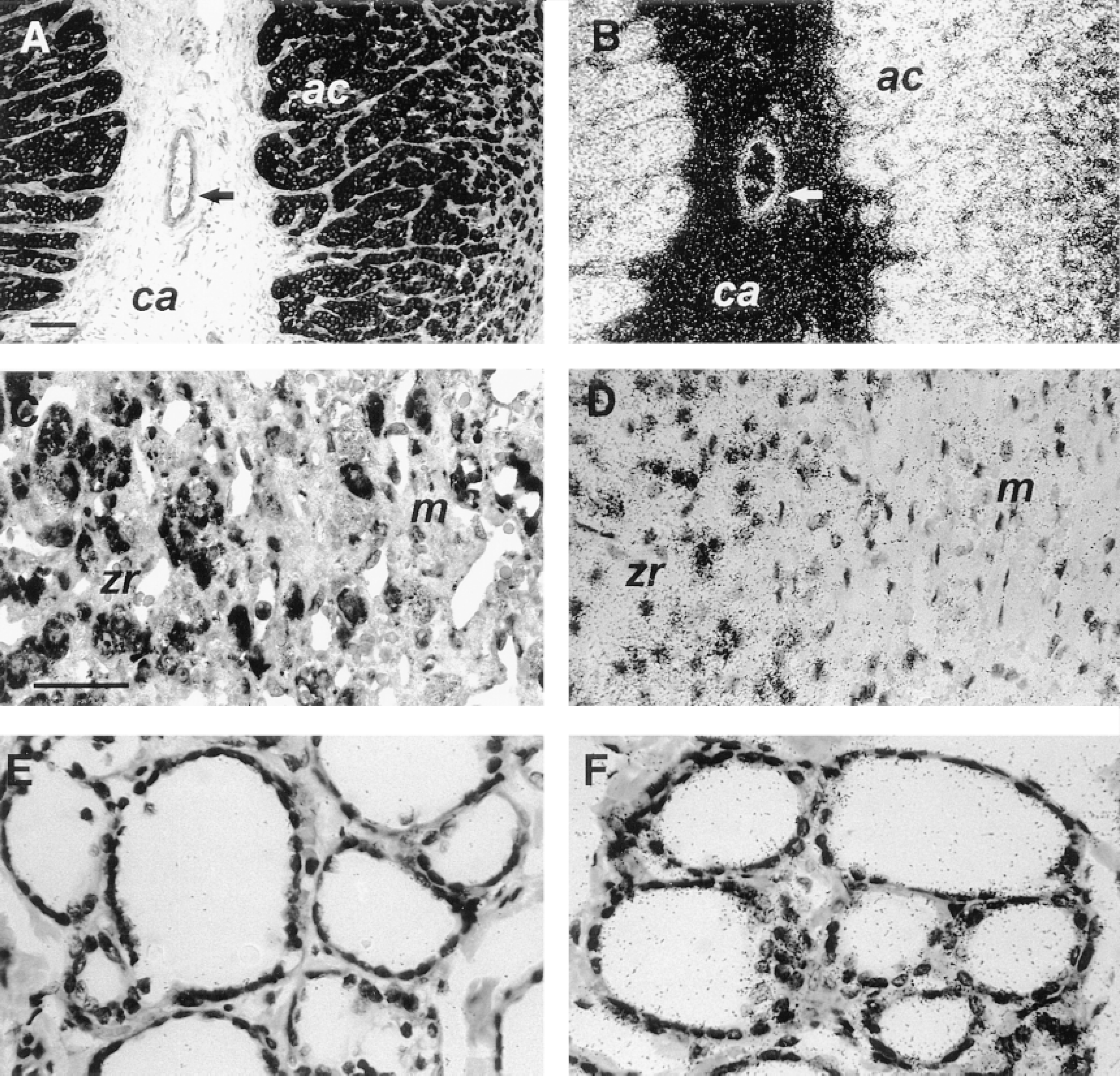
Human ECE-1 protein (A,C,E) and ECE-1 mRNA (B,D,F) in endocrine glands. Low-power brightfield (A) and darkfield (B) photomicrographs of serial sections of adrenal gland with high ECE-1 protein (A) and mRNA (B) levels in the adrenal cortex (ac) and no signal in the capsule (ca). Arrows point to a vessel in the capsule that contains both ECE-1 mRNA and protein. Bars = 100 μm. High-power brightfield photomicrograph of adrenal gland shows moderate ECE-1 protein (C) and mRNA (D) levels in zona reticularis (zr) and scattered staining in the medulla (m). In the thyroid, ECE-1 protein (E) and mRNA (F) are restricted to follicular epithelial cells. Bars = 50 μm.
Urinary System
The levels of ECE-1 mRNA and protein detected in the urinary system differed. In the kidney, there was strong staining for ECE-1 protein in the glomerulus (Figure 5D), whereas the staining intensity was weak to moderate in the distal and collecting tubules (Table 1). In contrast, ECE-1 mRNA levels were low in the glomerulus and undetectable in the proximal tubules (Figure 5D). No mRNA but moderate levels of ECE-1 were detected in the bladder epithelium, whereas there was no staining in the smooth muscle layers (Table 1).
Reproductive System
The hormone-secreting Leydig cells of the testis were strongly labeled for ECE-1 mRNA and protein, whereas the seminiferous tubules were only weakly labeled (Figures 4K and 5F). In the epididymis, moderate amounts of ECE-1 were detected in the epithelium, but ECE-1 was not detected in the smooth muscle cells (Table 1). In the prostate gland, ECE-1 was restricted to the glandular epithelium (Figure 5E). In the female reproductive system, there was a clear ECE-1 distribution pattern in the ovary. The follicular granulosa and theca cells were intensely labeled, whereas the surrounding stromal cells were only moderately or weakly labeled (Figures 3C and 3D). There was less ECE-1 in the oviduct than in the ovary, and it was restricted to the epithelium (Table 1). In this study, we examined breast tissue from nonpregnant nonlactating women only. Moderate levels of ECE-1 were detected in the duct and lobular epithelium, whereas the myoepithelial cells produced less ECE-1 (Figure 4I).
Endocrine Glands
ECE-1 labeling was strongest in endocrine tissue. In the adrenal gland, the strongest staining for ECE-1 was in the zone glomerulosa and fasciculata, with slightly less staining in the zona reticularis (Figures 6A and 6B). In the medulla, there was scattered but strong labeling (Figures 6C and 6D). The follicular epithelium of the thyroid was moderately labeled for ECE-1 (Figures 6E and 6F).
Lymphoid Tissues
In the thymus, there was weak ECE-1 labeling in the medulla, but no ECE-1 mRNA was detected in Hassall's corpuscules from the medullar region and the thymic cortex (Figure 4F). In the lymph nodes, high endothelial venules were strongly labeled for ECE-1 mRNA and protein, whereas sinus cells produced very little. The germinal centers in follicules were moderately labeled. In contrast, ECE-1 was absent from the T-cell region of the lymph nodes (Figures 4G and 4H), but small amounts were detected in the red and white pulp of the spleen (Table 1). The follicles of the lymphoid tissue of the tonsil were weakly labeled, whereas the surrounding lymphoid tissue was unstained. As in the esophagus, the basal cell layers of the stratified squamous epithelium were weakly labeled for ECE-1 mRNA and protein (Table 1).
Other Tissues
There was little or no ECE-1 mRNA and protein in the skin and its appendages, with the exception of the acinar cells and ducts of skin sweat glands, which were moderately stained for ECE-1 (Table 1). In contrast to white adipose tissue, brown adipose tissue had scattered patches of ECE-1 synthesis (Table 1).
Discussion
Endothelins (ETs) have a variety of biological effects in noncardiovascular tissues, and there is increasing evidence that ETs act locally rather than as circulating peptides. Endothelial cells in culture release ET-1, predominantly into the basolateral compartment (Wagner et al. 1992), and no ECE-1 was detected in plasma. The presence of ECE-1 in the same cell or in close proximity to its substrates is required for ETs to be effective local mediators. Clearly, the targeted disruption of ECE-1 gene showed that mature ETs must be produced at specific sites to influence development (Yanagisawa et al. 1998). Therefore, detailed knowledge of the cellular distribution of ECE-1 in all organs synthesizing preproETs is necessary and important to assess whether ETs exert any autocrine or paracrine effects on their target cells. In addition, knockout of ECE-1 revealed that the enzyme is involved in ET-1 and ET-3 generation. Because only few studies have concerned the localization of ET-3 peptide, mainly because of the nonavailibility of a specific antibody for mature ET-3 peptide, this study pinpoints the expression of the key component, ECE-1, involved in the generation of both ET-1 and ET-3 peptides.
We developed specific tools for detecting ECE-1 and its mRNA with high sensitivity in cells rather than merely in tissues. For immunohistochemistry, an anti-serum was raised against a synthetic peptide containing amino acids 473–489 of the extracellular part of human ECE-1, which recognizes the monomeric and dimeric forms of ECE-1 (Korth et al. 1997). We checked that this antiserum also reacted specifically with ECE-1 in formalin-fixed, paraffin-embedded specimens (Figures 1C and 1D). Another member of the ECE family, ECE-2, cloned from bovine adrenal cortex, is 59% identical in sequence to ECE-1 (Emoto and Yanagisawa 1995), so we needed to evaluate any crossreaction of ECE-1 antibodies with ECE-2. By preincubation of the antiserum with either the synthetic peptide containing amino acids 473–489 of ECE-1 or the synthetic peptide containing amino acids 502–518 of the corresponding sequence in ECE-2, we demonstrated that the 473-17-A ECE-1 antiserum does not recognize ECE-2. Furthermore, we investigated whether there was any crossreaction of anti-ECE-1 antiserum with the closest member of the zinc metalloprotease family, NEP, by immunostaining of serial sections with a commercially available anti-NEP antibody and the ECE-1 antiserum. These two sera gave different staining patterns. This demonstrates that anti-ECE-1 antiserum was specific for ECE-1 only, as previously shown by Western blotting (Korth et al. 1997). PEX, another close member of the zinc metalloprotease family with a sequence 37% identical to that of ECE-1 (Beck et al. 1997), has been studied in human fetal tissues and adult mice by RNase protection assay. It was present only in the calvaria, bone tissues, lung, and muscle of human fetuses and in lung and bone in adult mouse. Therefore, the 473-17-A ECE-1 antiserum recognizes specifically ECE-1 without any crossreaction for another ECE-like enzyme or for the closest member of zinc metalloprotease family, NEP. A riboprobe corresponding to nucleotides 304–1666 of human ECE-1a was used for in situ hybridization. The specificity of our riboprobe was demonstrated by the absence of background labeling by the sense probe of CHO cells overproducing ECE-1 (Figures 1A and 1B).
Three human ECE-1 isoforms (ECE-1a, ECE-1b, and ECE-1c) have been identified (Schmidt et al. 1994; Shimada et al. 1995; Schweizer et al. 1997). They are encoded by the same gene (Valdenaire et al. 1995). All isoforms are widely distributed in human tissues, but their relative mRNA abundances differ, with isoform c being predominant. However, no functional differences in terms of ECE activity have been reported. Because the three isoforms differ only at the N-terminal extremities of their cytoplasmic domains and their sequences in the area chosen for riboprobe transcription and for raising our ECE-1 antibody are identical, three types of ECE-1 mRNA and protein would have been detected in the human tissues studied.
There are known differences in the distribution patterns of ETs between rodent species and humans (see below), which led us to study ECE-1 expression in human tissues. This study aimed to (a) determine the distribution of ECE-1 in most tissues; (b) compare the distribution of ECE-1 mRNA with that of ECE-1 protein; (c) compare the level of ECE-1 produced in the cardiovascular system with that of other tissues; and (d) clarify whether the distribution of ECE-1 is the same as that of its substrates (bigETs) and their corresponding receptors (ET-A and ET-B).
The ECE-1 gene was expressed in diverse human tissues, suggesting a major biological role for the enzyme. ECE-1 was not strictly limited to endothelial cells in the organs examined but was also detected in mesenchymal and epithelial cells. In almost all organs, the cellular distributions of ECE-1 mRNA and ECE-1 protein were identical, demonstrating that ECE-1 mRNA synthesis and protein expression occur at the same site. This result is not surprising because ECE-1 is a membrane-anchored zinc metalloprotease and no soluble secreted form has yet been described that could potentially be detected in a cell compartment other than that in which mRNA was localized. However, the relative ECE-1 protein and mRNA contents differed in some epithelia. In the bladder, ECE-1 mRNA labeling was lower than that of protein staining, in contrast to the respiratory epithelium, in which mRNA and protein were present at equivalent intensities. This may reflect different stabilities of ECE-1 protein between tissues and suggests that there may be different functions of ECE-1 in different tissues.
The strongest staining was associated with the vascular endothelial cells of most organs examined. This is consistent with the work of Xu et al. (1994) and Takahashi et al. (1995), who studied bovine ECE-1 mRNA (Xu et al. 1994) and rat ECE-1 protein (Takahashi et al. 1995) levels in endothelial cells from various tissues (e.g., coronary artery, heart, pancreas, lung, liver, and kidney).
Although ECE-1 has been cloned from cultured human endothelial cells, ECE-1 mRNA and protein were only very recently detected for the first time in endothelial cells in human kidney sections by in situ hybridization and immunohistochemistry (Pupilli et al. 1997). ECE-1 was detected in the endothelial layer throughout the cortical and medullary vasculature. This study investigated other human organs and showed that endothelial expression is predominant in many organs of the human body and that there is little or no ECE-1 in the vascular smooth muscle cells and the adventitia. This is consistent with ET peptides being generated at the same site as their inactive precursors (preproETs) and with ETs having an autocrine/paracrine effect on the ET receptors of endothelial and vascular smooth muscle cells. ECE-1 is present throughout the vascular endothelium, whereas the ECE-1 staining intensity in the media is more variable, possibly because of phenotypic variability in the smooth muscle cells due to conditions such as hypertension or the early stages of atherosclerosis, not detectable by normal histological examination. This is consistent with the work of Minamino et al. (1997), who detected ECE-1 only in the neointimal smooth muscle cells of balloon-injured rat arteries, whereas, in uninjured arteries, ECE-1 was restricted to endothelial cells. They also detected ECE-1 in human smooth muscle cells and macrophages under pathological conditions, such as human coronary atherosclerotic lesions. Therefore, ECE-1 is a potential target for the treatment of atherosclerosis and angioplasty restenosis.
Strong staining intensity of ECE-1 was observed in a variety of epithelial tissues. As expected, epithelial cells of the respiratory system, such as those of trachea and main bronchus, were labeled. This is not surprising because ECE was initially purified from rat lung (Takahashi et al. 1993). Epithelia from the gastrointestinal tract (e.g., stomach), reproductive system (e.g., prostate), and thyroid also contained ECE-1, although to a lesser extent.
The reproductive system has among the highest staining intensity of ECE-1. In the testis, ECE-1 staining was higher in the Leydig cells than in the seminiferous tubules, which were only weakly stained. Maggi et al. (1995) detected ET-1 and its mRNA in Sertoli cells, whereas very few tubule and interstitial cells were stained. mRNA transcripts for both receptor subtypes were detected by Northern blotting with ET-A as the major subtype. Maggi et al. found that ET binding sites were concentrated in the spermatocytes and early spermatids, whereas the Sertoli cells had no such binding sites. Therefore, all the components of the ET system are presumably synthesized in the testis, but not all in the same cell. The enzyme ECE-1 is found mostly in Leydig cells, its product ET-1 in Sertoli cells, and the target cells for ET-1 are mainly developing germ cells in the seminiferous tubules. PreproET-1, ET-1, and ECE-1 have all been detected in the epithelial cells of the human epididymis (Peri et al. 1997). In this study, we extended findings over ECE-1 localization and detected moderate amounts of ECE-1 in the epithelium but not in the muscularis. These cells synthesize both classes of ET receptors (Peri et al. 1997), suggesting that ET-1 is one of the paracrine factors responsible for sperm progression through this organ. Epithelial ECE-1 may also convert some of the seminal bigET-1 into ET-1, which suggests that ETs also act as hormonal factors in human epididymis.
ECE-1 was also investigated in the female reproductive system. ECE-1 mRNA and protein labeling were strong in the granulosa and theca cells of the ovary. Magini et al. (1996) detected ET-1 and its mRNA in the granulosa cells, and ET-1 alone in follicular fluid. Therefore, at least some of the ET-1 produced in the granulosa cells is presumably secreted into the lumen of ovarian follicles. ET receptors have been identified in the blood vessels of the ovary and in the theca interna of ovulating follicles (Mancina et al. 1997). In contrast to the situation in the rat ovary, ET-B receptors were predominant in granulosa cells. There was thus a distinct species difference in the levels of synthesis of the members of the ET family. The high staining intensity for ECE-1 in theca cells in the absence of preproET-1 suggests that there may be another substrate for ECE-1 in this cell layer. The functions of ECE-1 and ET-1 in follicular development are unknown.
Strong staining intensity for ECE-1 was detected in the adrenal gland. There was an ECE-1 gradient accross the zones of the adrenal gland, with the zona glomerulosa having the highest staining. Autoradiography has shown that ET-A receptors are limited exclusively to the zona glomerulosa, whereas ET-B receptors are found throughout the adrenal cortex (Belloni et al. 1994). Davenport et al. (1996) found that ET peptides and receptors were differentially distributed in the human adrenal gland. ET-1 was the predominant mature isoform and was found mostly in the adrenal vasculature. Subtype-selective ligands showed the same differential distribution of ET receptors within human adrenal glands. The synthesis of all the components of the ET system in the zona glomerulosa, the proliferative layer involved in the growth of the entire adrenal cortex, suggests that the ET system is involved in this biological effect. This was recently demonstrated by Mazzochi et al. (1997), who found that ETs stimulate the proliferation of rat adrenal zona glomerulosa cells via ET-A receptors.
The use of a sensitive, specific RNA probe and a specific polyclonal antibody made it possible to map in detail the cellular distribution of ECE-1 mRNA and protein in the cells of a variety of human tissues. This approach could be used for retrospective analysis of a large number of paraffin-embedded autopsy and surgical archive specimens. ECE-1 mRNA and its protein were abundantly synthesized in all endothelial cells and in a number of nonvascular cell types, consistent with the ET system being mainly paracrine in nature. The high level of ECE-1 in endocrine cells (e.g., zona glomerulosa and zona fasciculata of the adrenal gland, granulosa and theca cells of the ovary, and Leydig cells of the testis), suggests that ECE-1 may be involved in other systems, such as the regulation of hormone secretion, rather than exclusively generating ET-1 from its precursor. This idea is supported by a recent study showing the hydrolysis of bradykinin by ECE-1 (Hoang and Turner 1997), suggesting a broader specificity for this enzyme than was previously recognized. The targeted inactivation of the ECE-1 gene in mice showed that although there was still ET-1 in plasma due to the cleavage of bigET-1 by ECE-2, it did not rescue the mutant developmental phenotype. This demonstrates that mature ETs must be produced at specific sites to influence development (Yanagisawa et al. 1998). Because ECE-1 has not been detected in plasma, identifying the organs and cell types that produce ECE-1 is critical to determine the site of action of future specific ECE-1 inhibitors, which could be used in new treatments for a number of cardiovascular diseases. Our observation that ECE-1 is also widely distributed in noncardiovascular tissue is of clinical interest because this will have to be taken into account when the potential therapeutic and side effects of ECE-1 inhibitors are considered.
Footnotes
Acknowledgements
Supported in part by the Ministère de la Recherche et de l'Enseignement (ACC-SV9). Dr P. Korth received funding from the Fondation pour la Recherche Medicale.
We would like to thank H. Wallenfels, M.-T. Morin, and F. Mongiat for technical expertise, Dr J.-M. LeMoullec for providing ECE-1 cDNA, and Dr J.-M. Gasc for helpful discussion.