
Abstract
We used a combination of morphologic and histochemical methods to demonstrate that rat calvaria-derived mesenchymal cells, RCJ 3.1C5.18, in culture progress through the differentiation pathway exhibited by chondrocytes in the endochondral growth plate. The cells were grown either as monolayer or suspension cultures. Subconfluent monolayer cultures did not express markers typical of chondrocyte phenotypes. However, after reaching confluency the cells formed nodules of chondrocytic cells separated by cartilage-appearing matrix and encapsulated by fibroblast-like cells. Suspension culture produced cell aggregates with similar characteristics. Matrix in both the nodules and aggregates stained for collagen Types II and XI and aggrecan, and some cells displayed a distinctive pericellular matrix that stained for Type X collagen. Mineralization was evident in older cultures. By electron microscopy, most cells in the aggregates appeared as typical chondrocytes. However, some larger cells were surrounded by a “mat” of matrix comprised of hexagonal arrays of dense nodules interconnected by a filamentous network. Immunogold localization confirmed the presence of collagen Type X in this matrix. Analysis of markers of chondrocyte differentiation and terminal differentiation over time showed that these markers were acquired sequentially over 2 weeks of culture. This model system will be useful to study the regulation of various steps in the chondrocyte differentiation pathway.
The growth plate is a dynamic tissue in which cartilage produced de novo is converted into growing bone, i.e., endochondral ossification (Horton 1993). The cells that occupy the growth plate participate in a developmental program that involves their sequential proliferation, maturation, and terminal differentiation. Defining the biology of normal bone growth and elucidating aberrations in various bone growth disorders requires an understanding of the regulation of chondrocyte differentiation. However, the regulatory mechanisms have been elusive, especially for the mammalian growth plate. One of the major problems has been difficulty in faithfully reproducing the terminal events of chondrocyte differentiation in vitro.
Several groups have described cell culture conditions that support terminal differentiation of avian, mainly chicken, chondrocytes. For example, the appearance of the most commonly accepted markers of terminal differentiation, collagen Type X and alkaline phosphatase, has been observed in agarose suspension cultures of chondrocytes that had been previously dedifferentiated by monolayer culture (Castagnola et al. 1986). Similar results have come from micromass cultures of limb bud mesenchymal cells (Ahrens et al. 1977). More recently, similar differentiation programs have been observed in mammalian cell culture systems. Rat chondrocytes in pellet culture were shown to develop morphological features very similar to those of the growth plate (Ballock and Reddi 1994). Alini et al. (1996) reported a serum-free culture system in which prehypertrophic chondrocytes from the fetal bovine growth plate terminally differentiated in the presence of thyroid hormones T3 and T4. These authors observed sequential expression of Type X collagen and alkaline phosphatase and mineralization of matrix.
We have taken a different approach using mesenchymal stem cells with the potential for chondrocyte differentiation. Such cells have been obtained from murine embryonic tissues and bone marrow stromal cells. One mouse cell line, C3H/10T1/2, can differentiate into myoblasts, adipocytes, chondrocytes, and osteoblasts under different conditions (Taylor and Jones 1979). However, terminal differentiation has not been described. Other mesenchymal cell lines with similar differentiation capacities have been isolated from rat bone marrow stromal cells (Owen 1985; Owen et al. 1987). The Aubin group has described a clonal cell population, RCJ 3.1, from fetal rat calvaria that behaves as progenitor cells in the differentiation of muscle, fat, cartilage, and bone (Grigoriadis et al. 1990, 1996). Recently, further subcloning has generated the RCJ 3.1C5.18 line, which is restricted to the cartilage differentiation pathway (Grigoriadis et al. 1996; Lowe et al. 1996; McDougall et al. 1996).
We report here that the RCJ 3.1C5.18 (C5.18) cell line expresses both differentiated and terminally differentiated chondrocyte phenotypes in a sequence that mimics that of growth plate chondrocytes. We also describe a new antibody directed against peptides derived from both the NC1 and NC2 domains of mouse Type X collagen, which have been used to monitor terminal differentiation.
Materials and Methods
Materials
Dexamethazone, β-glycerolphosphate, fluoroscein-conjugated secondary antibodies, phosphatase substrate 104, keyhole limpet hemocyanin, Aprotinin, and phenylmethylsulfonyl fluoride were obtained from Sigma Chemical (St Louis, MO). Fetal bovine serum, MEMα medium, and antibiotics were purchased from Gibco BRL (Gaithersburg, MD). Gold-conjugated secondary antibodies were purchased from Amersham (Arlington Heights, IL).
Antibodies
Two peptides, one derived from a sequence within the NC1 domain (KDEILYNRQQHYDPRSGI) and one from within NC2 (ERYQTPTGIKGPLASPKTQYF) of mouse Type X collagen, were synthesized. The amino terminal lysine in the NC1 peptide is noncoded and was used to enhance coupling of the peptide to carrier protein. Peptides were purified by reverse phase HPLC and coupled to keyhole limpet hemocyanin (Reichlin 1980). Polyclonal antisera, designated pXNC1 and pXNC2, were raised against these conjugated immunogens in New Zealand White rabbits using standard immunization protocols. Polyclonal antisera to Type XI collagen (Oxford et al. 1994) and aggrecan were provided by Nick Morris and Kurt Doege, respectively. The monoclonal II-II6B3 developed by Tom Linsenmayer was obtained from the Developmental Studies Hybridoma Bank maintained by Johns Hopkins University School of Medicine (Baltimore, MD) and the University of Iowa (Iowa City, IA), under contract from the NICHD. All animal handling was done according to institutional guidelines.
Cell Culture
The C5.18 cell line was maintained in MEMα medium supplemented with 15% fetal bovine serum and 10−7 M dexamethazone. Subconfluent cultures are routinely passed every 3.5 days at a plating density of 1.6 × 103 cells/cm2. Cells were cultured in six-well dishes (Costar) for Alcian blue and alkaline phosphatase assays and on LAB-TEK plastic chamber slides (Miles Scientific; Naperville, IL) for immunofluorescent staining. The differentiation of cartilage nodules was initiated when confluent monolayers were additionally supplemented with ascorbic acid (50 μg/ml) and β-glycerolphosphate (10 mM). Differentiating cultures were fed supplemented media every 3.5 days. These cultures were monitored over a total period of 14 days. For suspension culture freshly trypsinized cells were resuspended (5 × 104 cells/ml) in supplemented media and 10 ml distributed into 100 mm bacteriological dishes. Under these conditions the cells form small aggregates and proceed through a cartilage pathway similar to that described for cells cultured over agarose (McDougall et al. 1996).
Cell Layer and Tissue Staining
Immunofluorescent studies were performed with cells cultured on plastic chamber slides and aggregates generated in suspension culture. Mouse limbs and cell aggregates were fixed in ice-cold hexanes, suspended in Tissue-Tek embedding medium (Miles Laboratories), and sectioned in a Leitz 1720 cryostat. Tissue sections and cell layers were fixed in acetone and stained with antibodies as previously described (Sakai and Keene 1994). von Kossa staining for mineral is described elsewhere (von Kossa 1901).
Quantitation of Alcian Blue Staining and Alkaline Phosphatase Activity
Cartilage matrix deposition in cells cultured in six-well dishes was quantitated by Alcian blue staining. Cell layers were stained with Alcian blue (1% in 3% acetic acid) for 30 min, washed three times for 2 min in 3% acetic acid, rinsed once with water, and solubilized in 1% SDS. The absorbance at 605 nm was determined for triplicate samples.
For alkaline phosphatase activity, cell layers were extracted in 2 ml of lysis buffer (100 mM Tris-HCl, pH 9.0, 200 mM NaCl, 0.2% Nonidet P-40, 0.2% Triton X-100, 1 mM MgSO4, 1 mM phenylmethylsulfonyl fluoride and 10 μg/ml aprotinin) by rotating plates for 30 min at 4C. Reaction mixtures, containing 50 μl of extract, 200 μl buffer, and 250 μl of phosphatase substrate (1 mg/ml in 20% diethanolamine-HCl, pH 9.8), were incubated for 30 min at 37C and the absorbance at 405 nm was determined for triplicate samples.
Western Blot Analysis
Cell layers in six-well dishes were extracted with 1% hot SDS and protein concentrations determined using the DC protein assay (Bio-Rad; Richmond, CA). Samples (10 μg/lane) separated on SDS-PAGE gels were blotted onto PVDF membranes (Millipore; Bedford, MA) in 10 mM CAPS, pH 11.0, and blocked in 5% dry milk. All incubations were in PBS, 0.05% Tween-20 at room temperature. Blots were incubated in primary antibody (diluted 1:1000) for 2 hr, washed three times for 10 min, and continued in peroxidase-conjugated goat anti-rabbit secondary (1:5000; Amersham) for 1 hr. Following another wash the blots were developed in SuperSignal Substrate (Pierce Chemical; Rockford, IL) and chemiluminescence detected with REFLECTION autoradiography film (DuPont NEN; Boston, MA).
Electron Microscopy
Electron microscopic evaluation and immunogold labeling of monolayer cultures and cell aggregates followed previously published procedures (Sakai et al. 1986). Briefly, cultures were immunolabeled by immersion in chondroitinase (290 U/ml PBS), then in primary antibody, then in secondary gold conjugate, then fixed in 1.5% glutaraldehyde/1.5% paraformaldehyde containing 0.1% (w/v) tannic acid, followed by 1% OsO4. Finally, the cultures were dehydrated and embedded in Spurr's epoxy. Cultures prepared for ultrastructural evaluation were simply rinsed in serum-free medium and immersed directly in the fixatives listed above.
Results
Shown in Figure 1 are phase-contrast photographs of C5.18 cells at various stages of culture. As subconfluent monolayers, these cells displayed a flattened appearance typical of mesenchymal cells (Figure 1A). At confluence the cells exhibited a cuboidal morphology (Figure 1B). Within a week after confluence and starting treatment with β-glycerophosphate and ascorbic acid, the cultures became extensively nodulated (Figure 1C). When the cells were grown as suspension cultures, they formed aggregates within a few hours that became highly refractile as a result of mineralization after 2 weeks (Figure 1D, arrow).
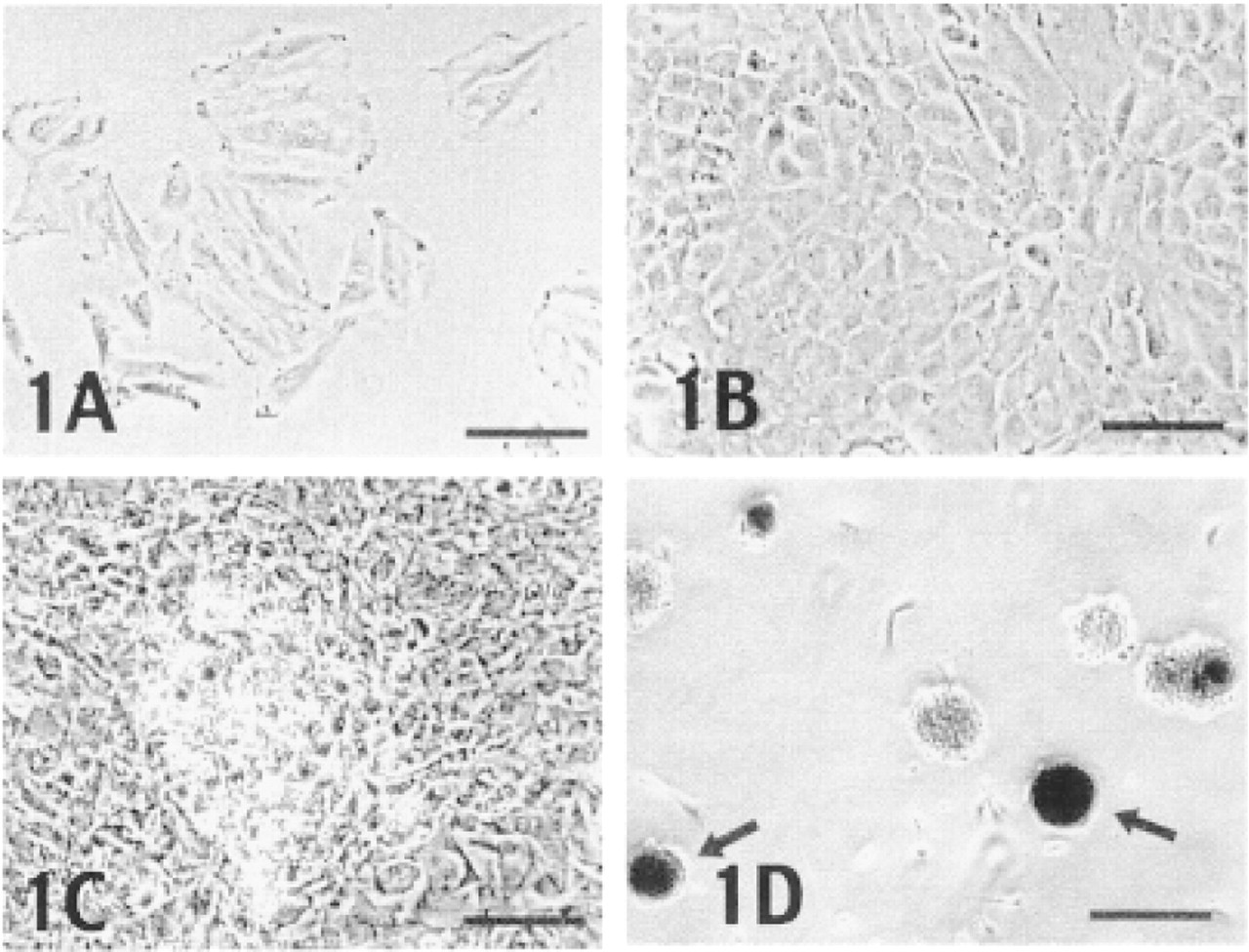
Phase-contrast microscopy of the C5.18 cell line at subconfluency (
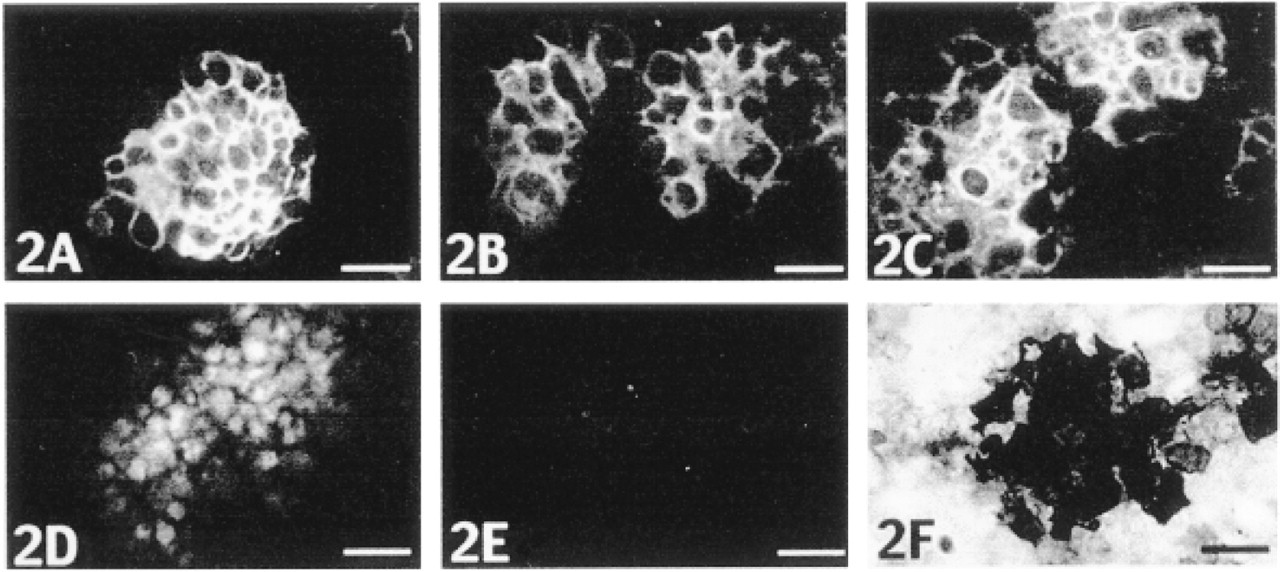
Immunostaining of nodules in monolayer culture with antibodies to Type II collagen (
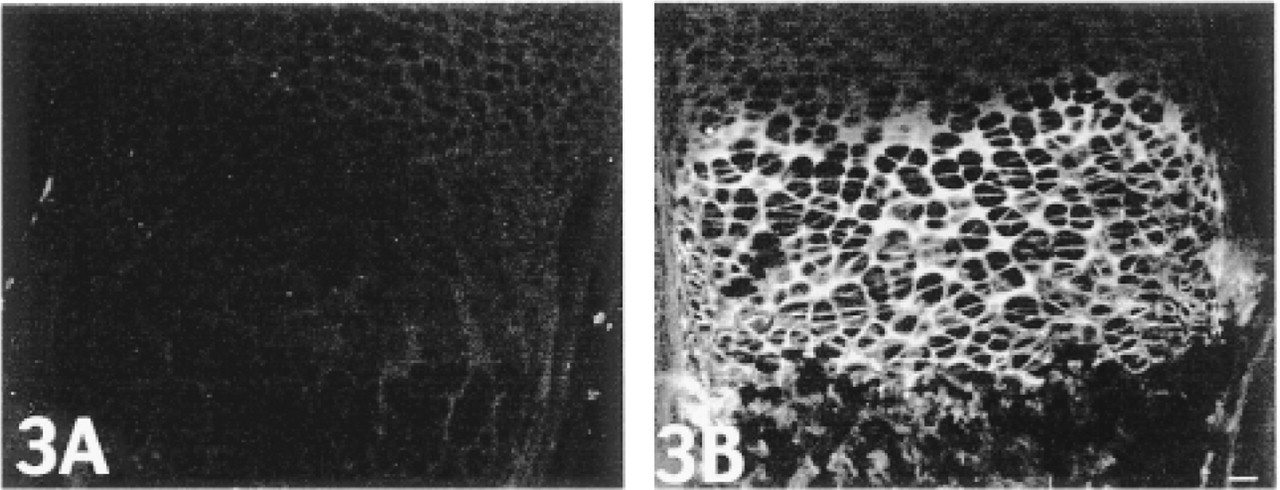
Type X collagen immunostaining with the pXNC2 antibody of tibial growth plates from 1-week-old mice.
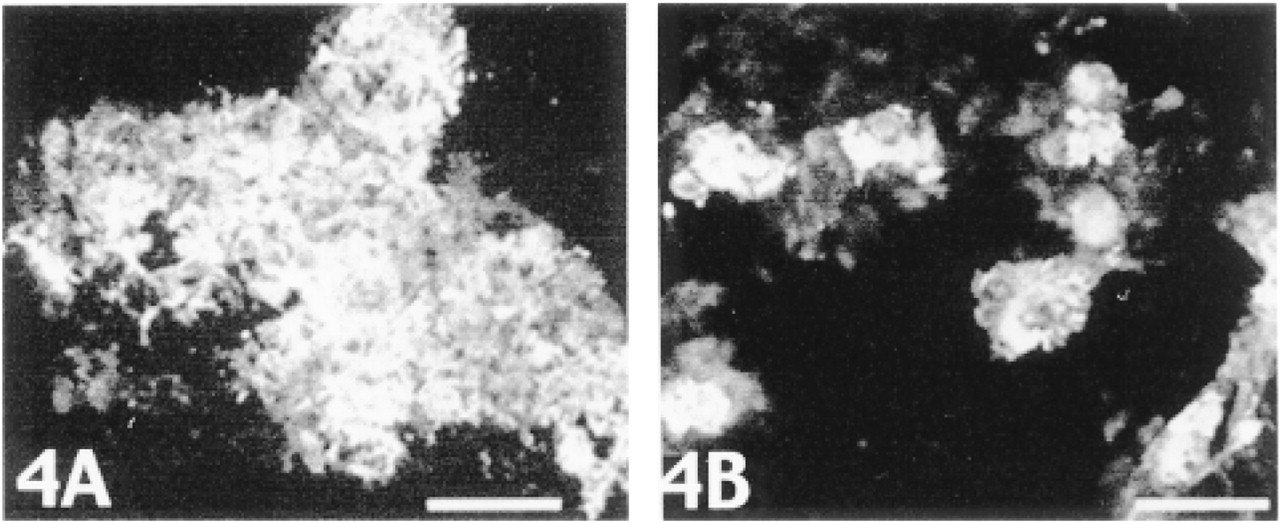
Immunostaining of aggregates grown in suspension culture for 2 weeks. Staining is extensive for Type II collagen (
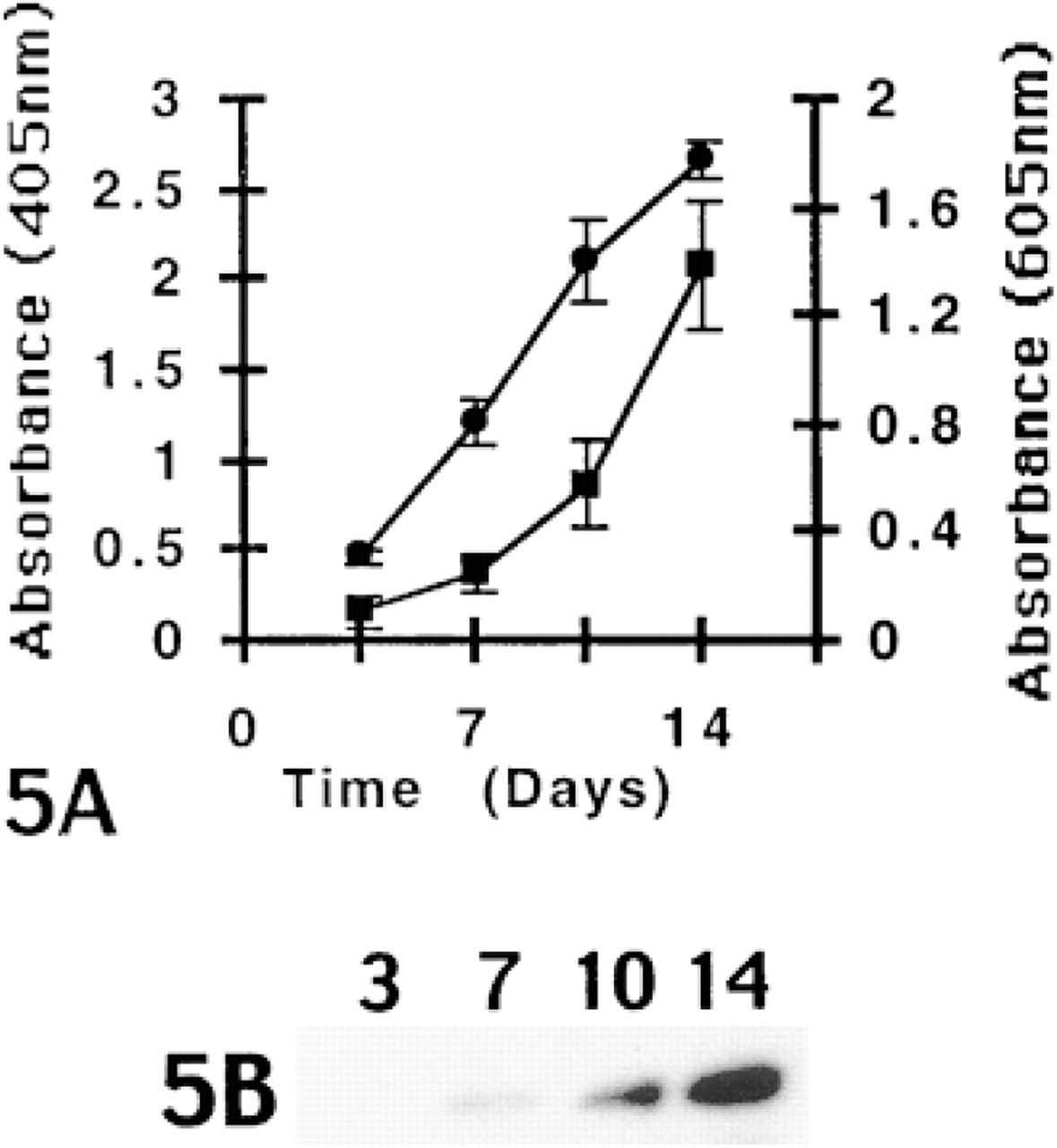
Temporal analysis of chondrocyte markers in monolayer cultures. (
The nodules, which formed in monolayer cultures, had a morphologic appearance similar to cartilaginous overgrowths that have been described in previous studies (Ahrens et al. 1977). The presence of cartilage matrix constituents was confirmed by immunofluorescent staining with antibodies to collagen Types II and XI and aggrecan (Figures 2A-2C). No staining was observed for these molecules in the monolayers between the nodules. Many of the cells in the nodules also stained positively for Type X collagen (Figure 2D) and exhibited mineralization (Figure 2F).
Because the antibodies raised against Type X collagen had not been characterized previously, long bones from newborn wild-type and Type X collagen null (Rosati et al. 1994) mice were immunostained. In the former, pXNC2 staining was limited to the hyper-trophic zones of long growth plates (Figure 3B) and secondary ossification centers (not shown). No staining was detected in other tissues or in growth plates from the null mice (Figure 3A). The pXNC1 antibody exhibited similar staining (not shown).
When cell aggregates grown in suspension culture were immunostained, the results were similar to the monolayer culture results. As shown in Figure 4, collagen Type II staining was widespread throughout the matrix (Figure 4A), whereas staining for collagen Type X was restricted to a few cells within the aggregates (Figure 4B).
The temporal sequence of chondrocytic differentiation and terminal differentiation is illustrated in Figure 5, which shows changes in Alcian blue staining, alkaline phosphatase activity, and Type X collagen expression. Starting with Day 3.5, Alcian blue staining increased linearly over the culture period. In comparison, there was a 3-4-day lag before a corresponding increase in alkaline phosphatase activity was detected. A similar time course was observed for the appearance of Type X collagen (Figure 5B). Together, these results show a distinct time course for chondrogenesis and terminal differentiation in the mesenchymal precursor cell cultures.

Ultrastructure of a 2-week-old cartilaginous nodule in monolayer culture. Both encapsulating fibroblast-like cells and chondrocytes are observed in a cross-section of the nodule (
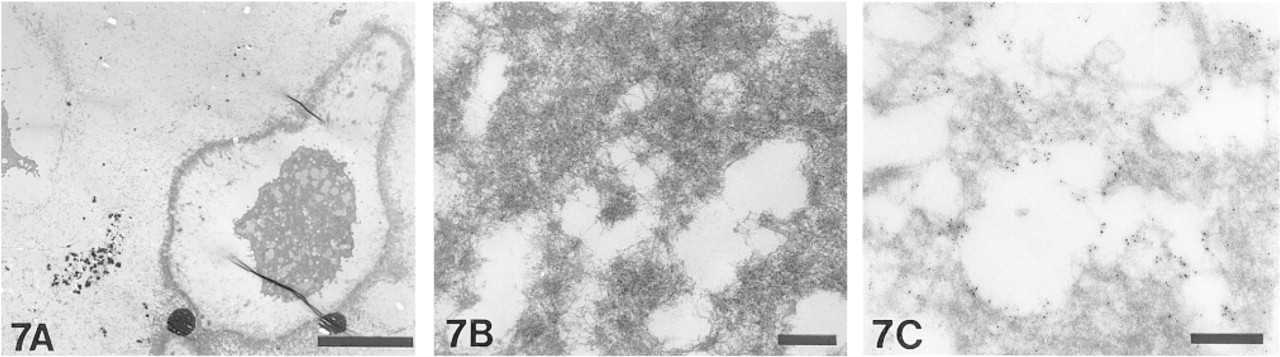
Electron micrograph of an aggregate from suspension culture. (
The ultrastructure of the nodules showed rounded chondrocyte-like cells surrounded by flattened fibroblast-like cells (Figure 6A). Collagen fibrils in the outer matrix were thick and were characteristic in structure and arrangement of fibrils composed of collagen Types I and III (Figure 6B). In contrast, the matrix separating the chondrocytic cells contained thinner fibrils typical of cartilage collagen fibrils composed of Types II, IX, and XI collagen (Figure 6C).
Electron microscopy of the aggregates demonstrated cells of different morphologies. Although most cells exhibited morphology typical of chondrocytes (Figure 7A, upper left), some were distinctly larger and were encapsulated by a pericellular “mat” of dense matrix (Figure 7A). At higher magnification, this pericellular mat was comprised of a hexagonal array of filamentous and globular elements (Figure 7B). Filaments are seen to span an interglobular distance of approximately 100 nm. Assuming that shrinkage of about 30% occurred during preparation for electron microscopy, this measurement is consistent with the 130-nm length of the Type X helix. The Type X collagen antibody directed immunogold labeling to this pericellular matrix, although the density of the matrix appeared to limit diffusion of the secondary gold conjugate into denser portions (Figure 7C).
Discussion
The differentiated and terminally differentiated chondrocyte phenotypes are unstable and difficult to reproduce in vitro. This most likely reflects the inability to provide cells external cues native to the growth plate in the artificial environment of cell culture. Nevertheless, our results suggest that C5.18 cells do progress through the differentiation pathway under the conditions described. The progression is not uniform, in that all of the cells do not express the identical phenotype at the same time of culture. Nevertheless, we believe that this culture system will provide a very useful tool for studying the transitions and the factors that influence the transitions which are relevant to understanding the regulation of the mammalian growth plate.
The organization of Type X collagen fibrils in the growth plate has been controversial. The observations that the closely related Type VIII collagen forms hexagonal arrays in tissues (Sawada et al. 1990) and that hexagonal lattice assemblies of Type X collagen also form in vitro (Kwan et al. 1991) has led to the suggestion that Type X collagen may form similar arrays in the growth plate. Our observations reveal similar filamentous assemblies in the pericellular matrix of putative hypertrophic chondrocytes in cell aggregates. Because the existence of Type X collagen arrays in tissue has not been documented, further studies will be needed to define their nature generated in vitro and confirm if they exist in vivo.
Footnotes
Acknowledgments
Supported by a Research Grant from the Shriners Hospital for Children. Electron Microscope Facilities were provided in part by the R. Blaine Bramble Medical Research Foundation and the Fred Meyer Charitable Trust.
We thank Dr Jane Aubin for the gift of the RCJ3.1C5.18 cell line. We gratefully acknowledge Jay Gambee for the preparation of synthetic peptides and the expert technical assistance of Cathy Ridgway.