
Abstract
I
We have encountered these problems in studies involving the detection of dystrophin in mouse muscles with murine MAbs. The dystrophin gene encodes 14-
Materials and Methods
Tissue Samples and Section Preparation
C57BL/10 and mdx mice aged from 3 months to 1 year were used. Muscles (five samples from five C57BL/10 mice and 25 samples from 20 mdx mice, including four muscle samples denervated 21 or 28 days previously) were dissected and snap-frozen in isopentane cooled by liquid nitrogen. Muscles from 3FTG β-galactosidase transgenic mice were also examined for nuclear β-galactosidase expression. Composite blocks of muscle samples from C57BL/10 and mdx mouse were used for initial tests. Sections 6 μm thick were cut onto 3-aminopropyltriethoxysilane (Sigma; Dorset, UK)-coated slides, dried, and stored at −70C until use.
Antibodies
MAb MANDYS8 (IgG2, 1:100 dilution; Sigma) and polyclonal rabbit Ab p6 (1:500 dilution, a kind gift from P. Strong, Neuromuscular Research Unit, Hammersmith Hospital, London, UK), both against dystrophin, were used for initial background and blocking tests on composite blocks of C57BL/10 and mdx mouse. A panel of six exon-specific MAbs to dystrophin, MANEX45A (IgG1, 1:4 dilution), MANEX50 (IgG1, 1:4 dilution), MANDYS101(IgG2b, 1:5 dilution), MANDYS110 (IgG1, 1:5 dilution), (all culture supernatants kindly supplied by G.E. Morris, MRIC Biochemistry Group, The North East Wales Institute, Wrexham, UK), NCL-DYS1 (IgG2b, 1:5 dilution; Novocastra, Newcastle Upon Tyne, UK), MANDRA1 (IgG1, 1:100 dilution; Sigma), as well as MAb GAL13 (IgG1, 1:100 dilution; Sigma) to β-galactosidase, D33 (IgG1, 1:50 dilution; DAKO, Cambridge, UK) to desmin were examined.
Immunohistochemistry
Sections were air-dried, hydrated in PBS, and incubated for 30 min with nonimmune normal serum (1:20 dilution) of the animals of the species in which the second layer Ab was raised, and then blocked with 1% H2O2 in PBS for 20 min (for immunoperoxidase staining only), followed by primary Ab for 1 hr at room temperature (RT).
For two-step detection methods, primary mouse Abs were detected with FITC-conjugated rabbit anti-mouse (RAM) Igs (0.4 mg/ml, 1:40 dilution; DAKO) or goat anti-mouse (GAM) Igs (0.8 mg/ml, 1:50 dilution; DAKO) or with horseradish peroxidase (HRP)-conjugated RAM Igs (1.3 mg/ml, 1:100 dilution; DAKO). Primary rabbit Ab p6 was detected with Texas red-conjugated donkey anti-rabbit (DAR) Igs (0.9 mg/ml, 1:200 dilution; Amersham Lifescence, Poole, UK) or HRPconjugated swine anti-rabbit (SAR) Igs (0.8 mg/ml, 1:200 dilution; DAKO).
For the three-step biotin-streptavidin detection method, sections were incubated with biotinylated RAM Igs (reacts with all mouse IgG subclasses, IgA and IgM, cross reactions with rat Igs and fetal calf serum have been removed; (0.6 mg/ml; 1:400 dilution; DAKO), or swine anti-rabbit (SAR) Igs (0.7 mg/ml, 1:500 dilution; DAKO) for 45 min followed by HRP- or Texas red-conjugated streptavidin (1:200 dilution; Amersham). Biotinylated RAM F(ab')2 (1:200; DAKO) was also used as secondary Ab. Enzyme activity of HRP was developed with 3,3'-diaminobenzidine tetrahydrochloride (DAB, 1 mg/ml PBS; Sigma) and 0.1% H2O2 for 4 min and the sections were counterstained with hematoxylin.
For double immunofluorescence staining, one of the MAbs and the polyclonal rabbit Ab p6 were used. Sections were incubated with both Abs simultaneously for 1 hr, followed by FITC-conjugated GAM Igs (1:50 dilution; DAKO) and Texas red-conjugated DAR Igs (1:200 dilution; Amersham) for 1 hr. All Abs were diluted in 3% bovine serum alMonoclonal Antibody on Homologous Tissues bumin (BSA; Sigma) in PBS and intervening washes of sections were carried out twice with PBS for 5 min unless otherwise stated. Negative controls were carried out by replacing the primary Ab with 3% BSA in PBS or by MAb NCL-DYS3 (IgG, 1:5 dilution; Novocastra), which is specific for human dystrophin and does not crossreact with mouse dystrophin.
Papain Digestion and Fab and Fc Preparations
Unlabeled RAM Igs (purified Ig fraction and reacts with all mouse IgG subclasses, IgA and IgM; 3.2 mg/ml; DAKO), RAM IgGs (purified IgG and reacts with mouse IgG, IgA, and IgM; 2.1 mg/ml; Sigma) and goat anti-mouse Igs (reacts with mouse IgG, IgA, and IgM; 1.6 mg/ml; DAKO) were incubated with papain (5% of Igs, w/w; Sigma), 20 mM
Fab and Fc fragments were separated from papain-digested Igs or IgGs by affinity chromatography using a HiTrip protein G affinity column (Pharmacia Biotech; Herts, UK) according to the manufacturer's instructions. Blocking efficiency of Fab or Fc was compared only with Fab-c from which the Fab and Fc were prepared.
Background Blocking
The following methods were examined for blocking efficiency: (a) washing with 1% Triton X-100 (Sigma) in PBS for 1 hr at RT before application of normal serum incubation; (b) incubation with fragments of papain-digested unlabeled anti-mouse Igs before application of primary Ab (see below); (c) blocking the binding of avidin to endogenous tissue biotin using a Biotin Blocking System (DAKO) according to the manufacturer's instructions; (d) blocking with mouse Fc receptor (FcR) blocking Ab, rat IgG2 anti-mouse CD16/CD32 MAb (Pharmingen; Cambridge, UK) at 10μg/ml for 1 hr before application of primary Ab; and (e) Incubation with purified normal rabbit Igs (DAKO) at a concentration of 2 mg/ml or with a papain digest of these Igs in place of normal rabbit serum. Papain-digested anti-mouse Igs and IgGs were used for blocking in the following forms and combinations: (a) Fab-c at a concentration ranging from 0.1 to 1 mg/ml; (b) Fab at a concentration of 0.1-0.5 mg/ml; (c) Fc at a concentration of 0.1 mg/ml; (d) Fab and Fc at the concentrations of 0.2 and 0.1 mg/ml respectively; and (e) Fab-c and Fc at the concentrations of 0.2 mg/ml and 0.1 mg/ml respectively. The initial incubation time ranged from 15, 30, 45 min to 1 hr at RT and 12 hr at 4C.
Results
Dystrophin was localized with rabbit anti-dystrophin Ab p6 at the sarcolemma of all muscle fibers in normal C57BL/10 mice. However, in the mdx mouse only a few sporadic fibers were positively stained (revertant fibers), all remaining fibers being negative for dystrophin when two- or three-step detection methods were used. When MAbs were used without blocking, background staining ranging from mild (five samples), to moderate (16 samples) to heavy (nine samples, including four denervated muscles) was observed. In sections with moderate background, a slightly stronger than background staining was suggestive of revertant fibers but could not be used as a reliable indicator (Figure 2A), whereas in sections showing heavy background revertant fibers were not distinguishable (Figure 3A). Therefore, revertant fibers could not be identified in most samples of mdx mouse with MAbs. Revertant fibers could be identified in samples with mild background only when the MAb-stained sections were viewed together with an adjacent section stained with polyclonal Ab p6. Background staining at similar intensity remained even when MAb NCL-DYS3 (specific to human dystrophin) was used or the primary Abs were omitted. That background was caused by the secondary anti-mouse Abs was supported by the fact that no background staining was observed when primary rabbit anti-dystrophin Ab was followed by biotinylated SAR Igs and peroxidase-conjugated streptavidin. A limited reduction in background was achieved when sections were prewashed with 1% Triton X-100 for 1 hr at RT with constant shaking. This reduction, however, was not sufficient for clear demonstration of revertant fibers in muscles with moderate and heavy background. Triton X-100 washes did not reduce specific staining for dystrophin, because signal intensity remained unchanged when polyclonal Ab p6 was applied to such washed sections.
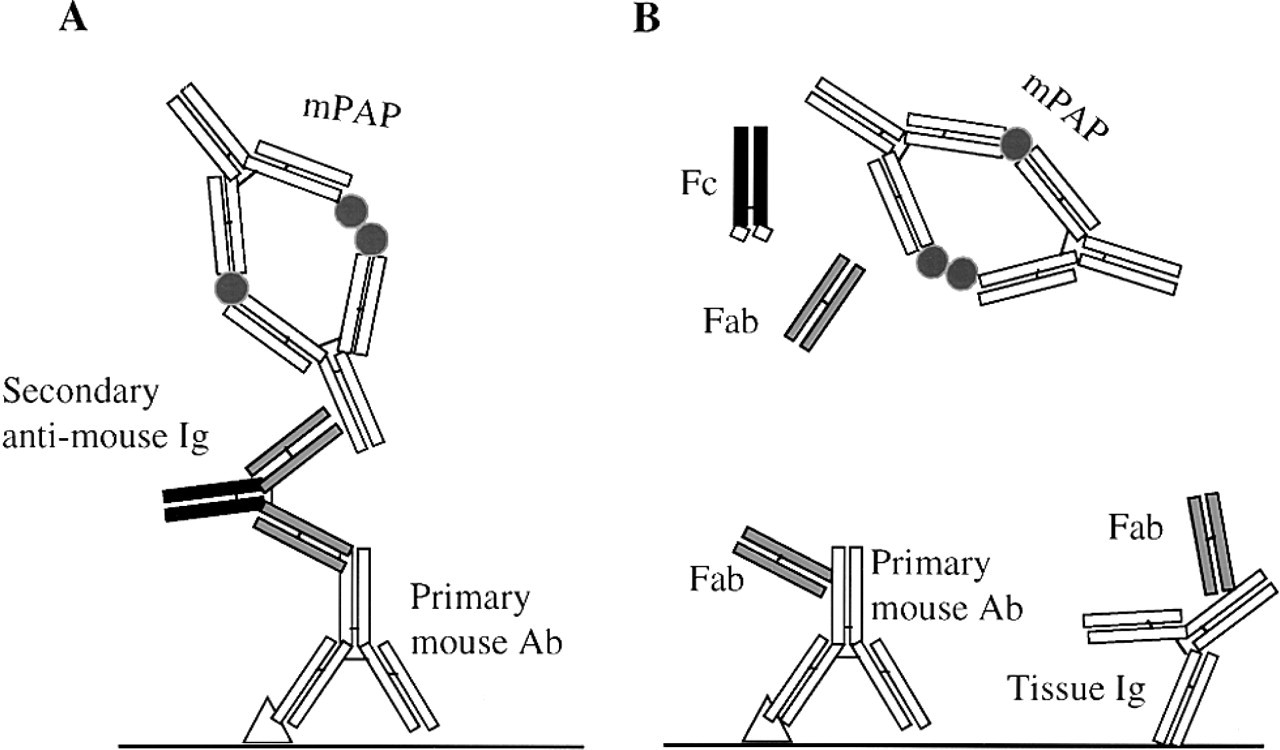
The principle for use of the mPAP method to determine the efficiency of papain digestion. (A) Normal mPAP immunohistochemistry using anti-mouse Igs as the bridge between the primary mouse Ab and the mPAP complex. Signal is produced specific for the primary mouse Ab. (B) mPAP immunohistochemistry using papain-digested anti-mouse Igs as second layer. Complete digestion with papain reduces the anti-mouse Igs to Fc and monomeric Fab fragments, thus abolishing their ability to bridge the primary mouse Ab and the mPAP complex. As a result, no signals for primary mouse Ab (or endogenous tissue Igs) are produced.
Muscle tissues are rich in endogenous biotin (Kirkeby et al. 1993). The level of background staining was lower with the two-step peroxidase or fluorescence detection system than with the biotin-streptavidin method, suggesting that endogenous avidin-biotin binding was involved in the high level of background. When sections were blocked with the Biotin Blocking System (DAKO) followed by the biotin-streptavidin detection system, a reduced level of background staining was observed in most samples but, again, not enough for a clear identification of dystrophin in revertant fibers with MAbs. Therefore, endogenous tissue biotin contributes to but is not the main cause of background staining in these muscles.
We then investigated the blocking efficiency of papain-digested rabbit anti-mouse Fab initially using the biotin-streptavidin method as a detection system. A significant reduction in background staining was observed when Fab of RAM Igs was used at 0.05-1 mg/ml with an optimal incubation time of 1 hr at RT (this duration was used thereafter unless otherwise stated). As a result, positive staining for dystrophin in revertant fibers was clearly identifiable with mAbs in 10 muscle samples, although weak background persisted (Figure 2B). These muscles showed intrinsically low to moderate background in the absence of blocking with Fab. However, in samples with innately heavy background, such as denervated muscles, it remained impossible to clearly identify revertant fibers, and Fab concentrations higher than 0.2 mg/ml did not reduce background staining further; rather, it augmented it. Failure to completely block the background could be due to the difference in binding specificity between secondary Ab and blocking Igs from which Fab was prepared. However, this proved not to be the case because a similar result was obtained when the blocking Fab was prepared from the same batch of RAM Igs that had been biotinylated as the secondary Ab.
Background staining, however, was almost eliminated in all muscle samples that showed low to moderate background (21 of 30 samples) when papaindigested Fab-c, containing both Fab and Fc of RAM Igs or IgGs, was used at concentrations between 0.1 and 0.5 mg/ml, being optimal at 0.2 mg/ml. This is shown in Figure 2C, where dystrophin in revertant fibers was unequivocally demonstrated with no background staining. In addition, this blocking treatment, when applied to muscles with heavy background, revealed some revertant fibers, although visible background persisted (Figure 3B). This result suggested that Fc of the RAM Igs was involved in the elevated background staining, possibly due to binding to endogenous mouse tissue Fc receptors or other tissue components. Because Fab-c at a concentration higher than 0.2 mg/ml was associated with increasing background, we used the Fc fragment alone or together with Fab-c for blocking. Fc of RAM IgGs alone reduced background staining to a greater extent than Fab alone, to a level similar to that with Fab-c in most muscle samples. When Fc (0.1 mg/ml) was used together with Fab-c (0.2 mg/ml) for blocking on sections with heavy intrinsic background, further reduction of background was achieved, below the level that could be attained with Fab-c blocking alone.
The conclusion that background staining was partly caused by binding of Fc fragments of secondary Ab to mouse tissue components was further supported by the result obtained using F(ab')2 as the secondary Ab. Muscle sections blocked with Fab-c/Fc and then visualized with biotinylated RAM F(ab')2 followed by Texas red or HRP-conjugated streptavidin showed a complete elimination of background in moderate background specimens. Even in the intrinsically high-background denervated muscles, background was barely visible with this regimen (Figure 3C) and, in combination with the three-step biotin-streptavidin amplification, allowed clear identification of staining for dystrophin in revertant fibers in all mdx muscle samples.
An unequivocal identification of nuclear antigen in muscle fibers was also possible with this system. This is shown in Figure 4 for detection of β-galactosidase in peripherally localized muscle nuclei.
The effective blocking with Fc fragments of antimouse Igs raised the possibility that a similar blocking effect could be obtained by incubation with high concentrations of normal rabbit Igs. This, however, proved not to be the case. Sections incubated with normal rabbit serum at 1:1 dilution, or with purified nonimmune rabbit Igs at up to 2 mg/ml or its crude papain digests, produced no noticeable reduction of background. To see whether mouse Ig FcRs were responsible for the binding of the Fc fragment of the secondary anti-mouse Abs, sections were incubated with rat anti-mouse CD16/CD32 MAb, which blocks nonantigen-specific binding of Abs to mouse Fcγ II/III receptor. Reduction in background was barely noticeable, suggesting that Fc of the secondary Ab binds to mouse tissue components other than the FcRs recognized by the Ab. When blocking with Fab-c/Fc at a concentration of 0.2 mg/0.1 mg/ml was applied to the two-step detection system with HRP- or FITC-labeled RAM Igs, specific signal for dystrophin and desmin, as expected, was slightly weaker than with the biotin-streptavidin detection system, with background negligible in all muscle samples.
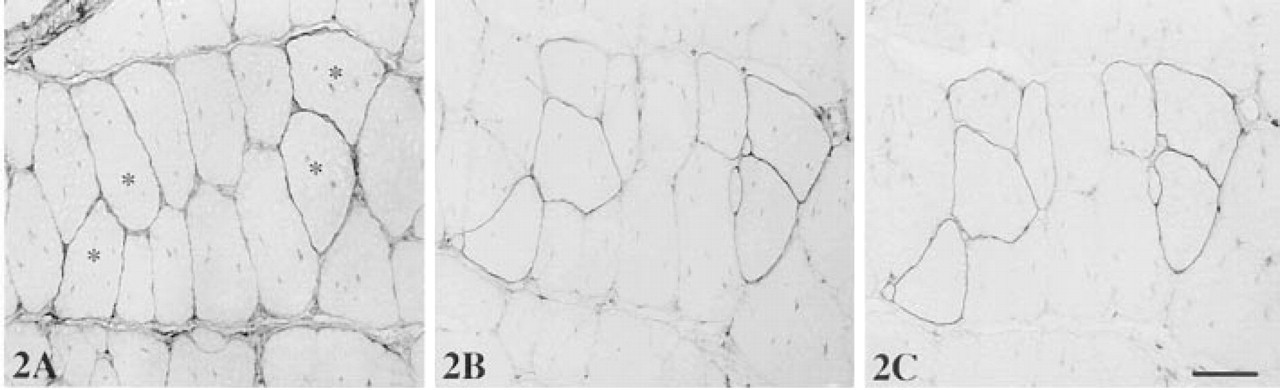
Three-step immunoperoxidase detection of dystrophin in mdx mouse muscle with MAb MANDYS8 and biotinylated RAM Igs as a second layer. (
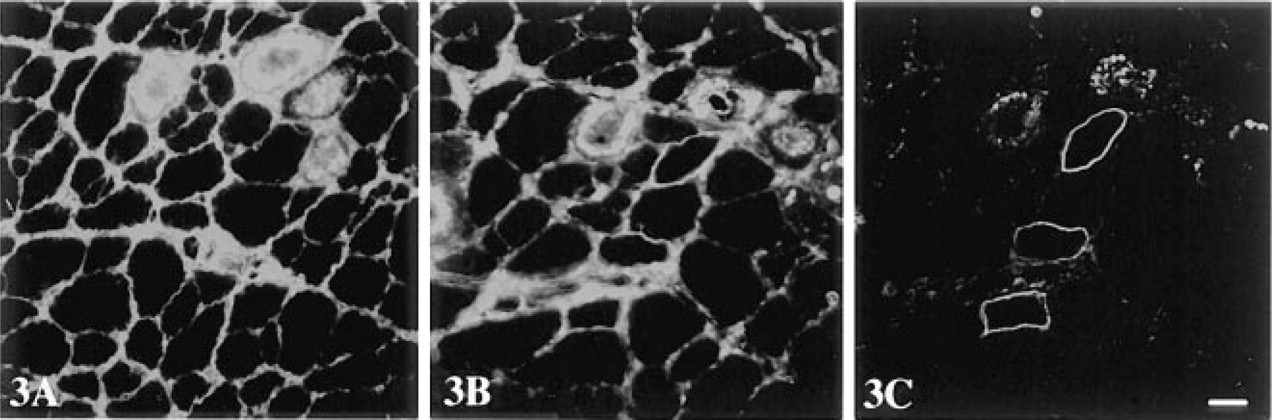
Three-step immunofluorescence (streptavidin-Texas red) detection of dystrophin on adjacent sections of denervated mdx mouse muscle with MAb MANDYS8. (
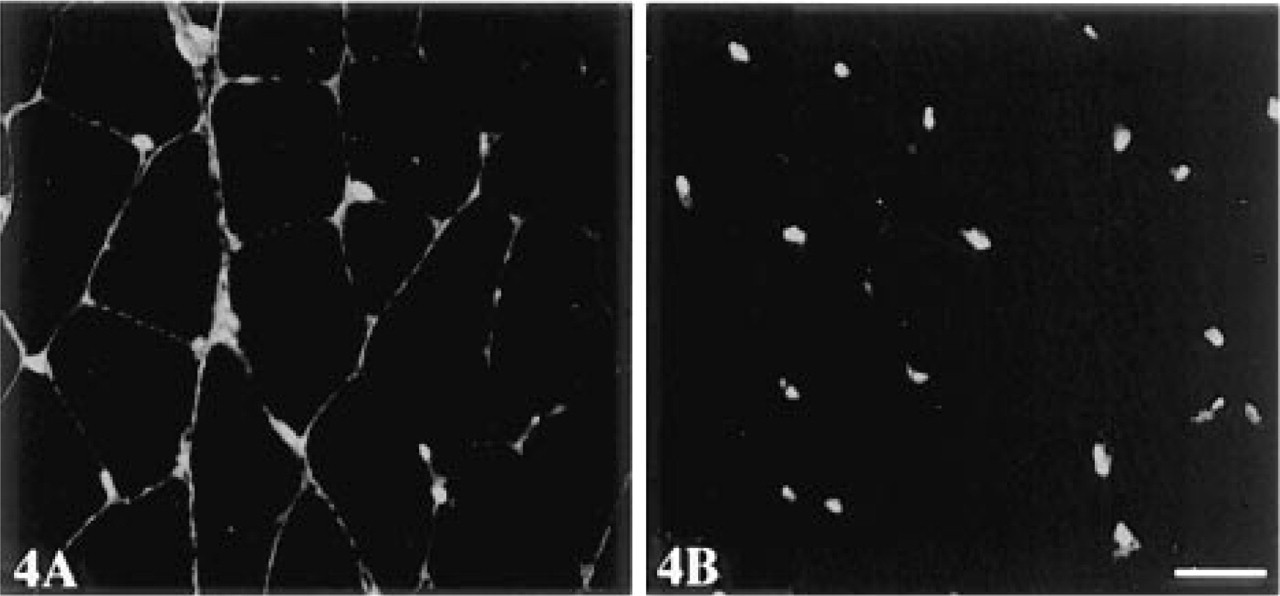
Three-step immunofluorescence (streptavidin-Texas red) detection of β-galactosidase in 3FTG mouse muscle with MAb GAL13. (
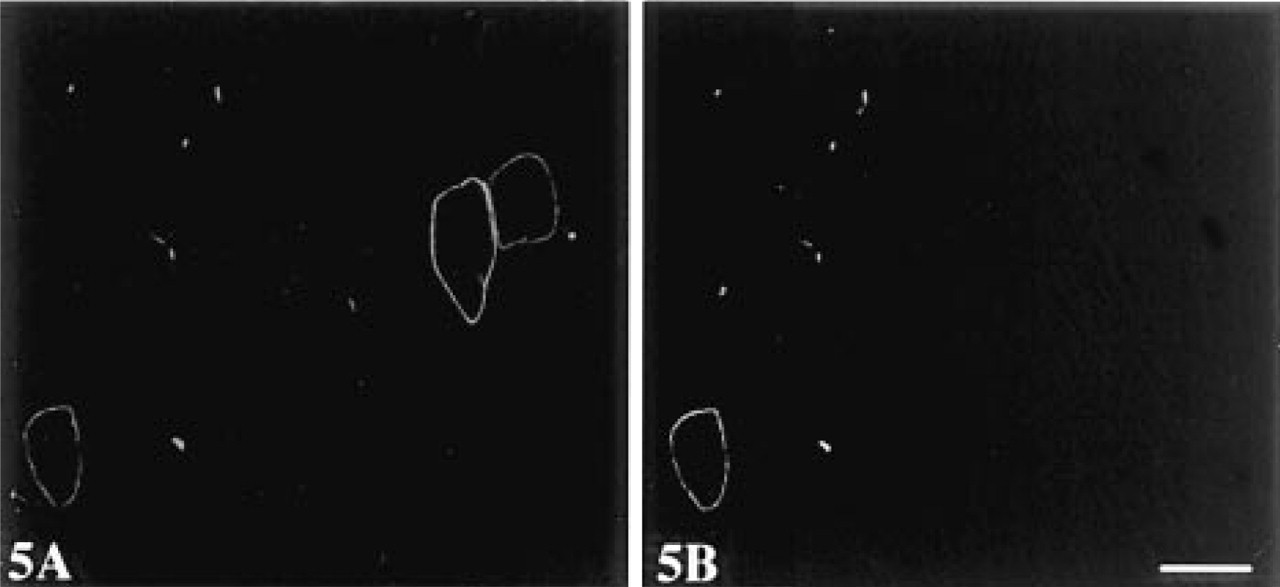
Immunofluorescence double labeling of dystrophin with MAb MANDYS8 (recognizes exons 31-32) detected with FITC-conjugated GAM Igs (A) and polyclonal Ab p6 (recognizes exons 57-60) detected with Texas red-conjugated DAR Igs (
To examine whether blocking with Fab-c was also applicable to double fluorescent staining, sections were blocked with Fab-c of GAM Igs at a concentration of 0.2 mg/ml and then incubated with one MAb to dystrophin or desmin and polyclonal rabbit Ab p6 to dystrophin followed by FITC-conjugated GAM Igs and Texas red-conjugated DAR Igs. Signals specific for each Ab were clearly demonstrated without interference between them. As shown in Figure 5, double staining with exon-specific Abs P6 and MANDYS8 identified revertant fibers expressing dystrophin with variation in exon composition on the same section. Whereas dystrophin expressed in almost all revertant fibers was stained by P6 to the C-terminal region, exon 31 to 32 detected by MANDYS8 was lost in a large proportion of revertant fibers.
Discussion
The background staining found when murine MAbs were applied to mouse tissues and detected by indirect methods was considered to be largely due to the presence of tissue Igs homologous with the primary Ab. If this were so, it should be possible to block the binding of secondary Ab to the tissue Igs by prior occupation of the binding sites with an Fab monomer derived from unlabeled secondary Ab. Nielsen et al. (1987) reported that background staining could be eliminated when human IgM was used as primary Ab on human tissues by preincubation of sections with monomeric Fab of unlabeled rabbit anti-human IgM and when the same batch of labeled rabbit anti-human IgM was used to detect the primary Ab. However, when the primary Ab was human IgG, only limited reduction in background was achieved by the same strategy. This difference may be attributable to the low tissue concentration of IgM such that background staining is often negligible, even without blocking, when a classspecific secondary Ab is used. The limitation of Fab blocking led to efforts to develop alternative methods for application of Ab on homologous tissues (Lewis Carl et al. 1993; Hierck et al. 1994). Hierck et al. (1994) reported a single-step indirect detection method in which mouse MAb and labeled anti-mouse secondary Ab were allowed to form complexes in solution followed by blocking of the unoccupied paratopes of the secondary Ab with Igs in normal mouse serum. The whole reaction mixture was then applied to mouse tissue sections. However, this method suffers from poor accessibility of the large Ab-Ab complexes to tissue antigens and thus from low sensitivity. Furthermore, the ratio for primary and secondary Abs must be optimized individually for each set of Abs, making it more difficult to obtain consistent and comparable results when different batches of Ab are used.
Our present investigation showed that failure to develop a satisfactory blocking method is due to the fact that background staining is not caused by a single factor when a homologous Ab is used. We found that endogenous peroxidase activity and the presence of endogenous tissue biotin and Igs all played some part but, surprisingly, that one major effect was the binding of the secondary Ab through its Fc fragment to tissue components. This was, in fact, consistent with the previous result (Nielsen et al. 1987) that failed to achieve satisfactory blocking with Fab of the same batch of unlabeled IgG, suggesting a possible binding of the Ab through sites other than Fab. Binding of Fc does not appear to be through the non-polymorphic FcR II/III receptor of endogenous mouse Igs, because blocking with Ab to the FcRs did not significantly reduce background. Interestingly, this binding cannot be blocked by high levels of normal rabbit Igs or its papain digests but can be effectively inhibited by the Fc fragment of anti-mouse Igs. This suggests that Fc of anti-mouse Igs has higher binding affinity than Fc of nonimmune serum of the same species, with the implication that immune rabbit serum contains a different spectrum of Ig isotypes from nonimmune serum, with Fc of greater nonspecific binding capacity to mouse tissue components. There is no particular reason why this should apply only to mouse tissues, and this phenomenon of nonspecific binding of Fc of immune secondary serum may underlie sporadic high background staining encountered when the primary Ab is not of the same species as the tissue being stained or when a polyclonal primary Ab is used. It is clear that background staining in the system studied above is multifactorial, and only limited reduction in background is achieved by blocking tissue sections for biotin binding or with Fab individually. Complete blocking was achieved in samples with moderate to heavy background only when sections were blocked against binding of both Fab and Fc of secondary Ab to tissue Igs and other components. The use of a crude papain digest of unlabeled secondary Ab for blocking provides a simple preparation that may provide Fab with strong antigen binding efficiency, which may possibly be attenuated during purification procedures. However, the most complete blocking was achieved by addition of extra Fc to the crude papain digest. In some tissues, blocking the binding of endogenous biotin or peroxidase activity may also be required.
In summary, our study shows that background staining encountered in the application of Ab to homologous tissues and detected by indirect immunohistochemistry mainly results from the binding of both Fab and Fc of secondary Abs to tissue Igs and other components. A simple and efficient blocking strategy was established that employs the unpurified papaindigested secondary anti-mouse Igs enriched with the Fc fragment of the same Igs. This, in combination with the biotin-streptavidin immunohistochemical detection system, effectively eliminates heavy background caused by the secondary Ab while providing maximal signal amplification, thus allowing detection of antigens expressed at low level with homologous Abs. Double labeling can be achieved with the blocking without signal interference. The principle and system can be adapted for wider applications in which Abs of other species can be used on homologous tissues and perhaps, in some instances, when unexplained high background staining is found with Ab and tissues of different species.
Footnotes
Acknowledgements
Supported by an MRC program grant and the Leopold Muller Bequest.
We are indebted to Professor Glenn Morris (MRIC Biochemistry Group, The North East Wales Institute, Wrexham, UK) and Dr Peter Strong (Neuromuscular Research Unit, Hammersmith Hospital) for Abs used in this work and to Drs Jennifer Morgan and Christian Pastoret, who kindly provided tissues from mdx and c57 mice.