
Abstract
D
Since the first descriptions of the DGC based on biochemical data (Ervasti et al. 1990; Yoshida and Ozawa 1990), a number of theoretical models of the structure of the DGC have been published in which dystrophin has usually been depicted as a dimer (Koenig and Kunkel 1990; Ervasti and Campbell 1993; Suzuki et al. 1994), occasionally as a tetramer (Ahn and Kunkel 1993) and, more recently, as a monomer (Winder et al. 1995; Campbell and Crosbie 1996). With the exception of the early model of dystrophin proposed by Koenig and Kunkel (1990), these representations have been linear one-dimensional arrays without a clear indication of how the complex may be assembled in a higher-order two- or three-dimensional network. The majority of the ultrastructural studies have, to date, been performed on sectioned material which, although confirming some of the essentials of the DGC organization, view it only in essentially one dimension (Watkins et al. 1988,1997; Cullen et al. 1990,1994,1996; Cullen and Watkins, 1993; Inoue et al. 1996; Wakayama et al. 1994,1995,1996). Wakayama's group has sought to expand our view of dystrophin and its associated proteins by carrying out immunolabeling combined with freeze-etching of the plasma membrane and the membrane cytoskeleton (Wakayama and Shibuya 1991; Wakayama et al. 1993). However, the efficiency of labeling with these techniques appears fairly low, with only a proportion of dystrophin molecules being labeled compared with the theoretical number available (Cullen et al. 1990), thereby making it difficult to distinguish any underlying pattern to the distribution.
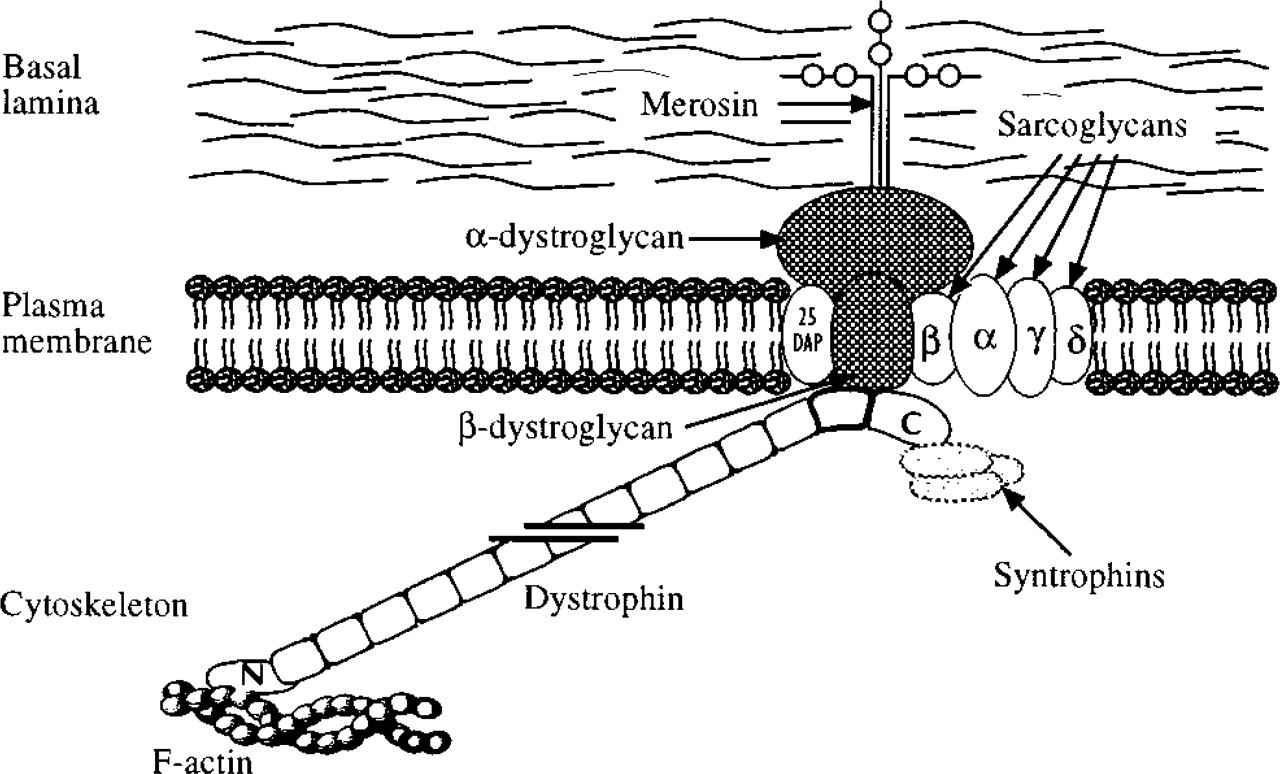
Diagrammatic representation of the organization of dystrophin and its associated proteins at the plasma membrane of skeletal muscle. The dystrophin rod domain, consisting of 24 coiled coil repeats, is shown truncated for ease of representation.
We have taken a different approach to obtaining a face-on view of the DGC by using immunogold labeling in combination with the freeze-fracture cytochemical technique, fracture-label (Pinto da Silva et al. 1986; reviewed in Severs 1995). The unique high-resolution single-and double-label views provided by this approach were combined with EM immunolabeling of cryosections and confocal microscopy to obtain a broader view of dystrophin and β-dystroglycan distribution and organization in skeletal muscle. We have recently successfully used similar approaches to examine and compare the distribution of dystrophin and vinculin (as a costamere marker) in cardiac muscle and have shown that in rat heart, at least, dystrophin is not exclusively a component of the costameres (Stevenson et al. 1997). In this study we examine how dystrophin and β-dystroglycan are distributed in rat skeletal muscle and consider the implications for the organization of the DGC. If the models of the DGC based on biochemical data are correct, we would predict that the patterns of labeling of the carboxy termini of dystrophin and β-dystroglycan will be essentially indistinguishable and that the two sites should exhibit extensive co-localization.
Materials and Methods
Tissue
Adult female rats (160-200 g) were stunned and sacrificed by cervical dislocation. The external digitorum longus (EDL) and soleus muscles were quickly removed and immersed in fixative while pinned, slightly stretched, to dental wax. The primary fixative was 2% paraformaldehyde plus 0.01% glutaraldehyde in PBS at pH 7.4. For both fracture-label procedures and immunolabeling after cryosectioning, the muscles were cut into blocks approximately 1 × 1 × 1.5 mm after 1 hr and fixed for a further hour. For fracture-label procedures, the blocks were cryoprotected with 30% glycerol (in PBS buffer) for 2 hr before freezing. For immunolabeling after cryosectioning, the blocks were cryoprotected with 2.3 M sucrose for 2 hr or overnight before freezing.
Antibodies
A polyclonal antibody, P1583, to the last 17 amino acids of dystrophin was a gift from Dr. Henry Klamut (Ontario Cancer Institute, Toronto) and was raised by synthesizing the peptide (SSRGRNTPGPMREDIM) and conjugating this to BSA or keyhole limpet hemocyanin. The antibody against the peptide was raised in rabbit and purified using affinity chromatography on CNBr-Sepharose 4B columns as previously described (Zubrzycka-Gaarn et al. 1988). The monoclonal antibody to β-dystroglycan, 43DAG/8D5, was a gift from Dr. Louise Anderson and was raised against a synthetic peptide representing 15 (PKNMTPYRSPPPYVP) of the last 16 amino acids at the C terminus of the published dystroglycan sequence (Ibrahimov-Beskrovnaya et al. 1992) conjugated to keyhole limpet hemocyanin. The fusion partner was the myeloma cell line X63.Ag8.653 and the MAbs were screened for reactivity on sections and blots of human, rat, mouse, dog, and chicken skeletal muscle.
Immunolabeling for Fracture-label
The glycerinated blocks of muscle were rapidly frozen by plunging them individually into liquid nitrogen slush (liquid nitrogen cooled to its melting point) and were then crushed under liquid nitrogen with a brass rod rotated in a copper well. Freeze-fracturing in this way generates fracture planes through the sample that preferentially follow the membrane planes, as in conventional freeze-fracture electron microscopy (Pinto da Silva et al. 1981a,b,1986; Severs 1995). The crushed (fractured) specimens were then allowed to thaw in the presence of pre-cooled fixative in 30% glycerol. The thawed specimens were rinsed in 30% glycerol to remove excess fixative, then deglycerinated by passage through 1 mmol/liter glycylglycine in 30% glycerol for 5 min, followed by pure 1 mmol/liter glycylglycine for a further 5 min.
For single labeling, the muscle fragments were incubated overnight in the primary antibody followed, after washing, by the appropriate secondary goat anti-rabbit or anti-mouse antibody conjugated with 10-nm colloidal gold for 1 hr. The anti-dystrophin antibody was used at a dilution of 1:1000 and the anti-β-dystroglycan at 1:50. After further washing, the specimens were postfixed in 2% paraformaldehyde plus 0.01% glutaraldehyde for 30 min. The larger fragments of the fractured muscle were then separated from the smaller pieces and prepared for platinum-carbon replication by partial dehydration (to 70% ethanol), air-drying, and mounting on the stage of a Balzers BAF 400T unit. Replicas were made by evaporation of platinum and carbon as in standard freeze-fracture, but at ambient temperature (Pinto da Silva et al. 1981a), carefully cleaned in sodium hypochlorite, collected on high transmission grids, and viewed in a JEOL 1200EX electron microscope.
The remaining smaller fragments of fractured muscle were prepared for sectioning. They were postfixed in 2% osmium tetroxide, dehydrated through a graded series of alcohols, and embedded in Araldite. Semithin and ultrathin sections were cut at right angles to the fracture plane using a Reichert Ultracut E microtome.
Double Labeling and Controls
In double labeling experiments the two proteins were labeled serially. A typical sequence would be as follows: (a) polyclonal antibody to dystrophin; (b) goat anti-rabbit conjugated to 10-nm gold; (c) MAb to β-dystroglycan; (d) goat anti-mouse conjugated to 5-nm gold. To control against steric hindrance of one conjugate by the other, the labeling sequence was repeated with the monoclonal preceding the polyclonal, and to control against the positioning being affected by the size of the gold conjugate the sequence was repeated with the secondary antibodies conjugated to the alternative size of gold. In some experiments, instead of a secondary goat anti-mouse conjugate, a biotinylated anti-mouse and streptavidin-gold system was used. Control experiments were also carried out in which dystrophin and β-dystroglycan were double labeled with an irrelevant non-co-localizing protein (myosin). Other controls run in parallel were (a) omission of primary antibodies and (b) a single primary with both secondaries. Cross-fractured cells in positively labeled samples served as internal controls.
(
Immunolabeling After Cryosectioning
Our methods of immunolabeling cryosections have been extensively documented in previous publications (Cullen et al. 1990,1994,1996). Briefly, sections were cut at a thickness of 80 nm at a block temperature of −90C and a knife temperature of −110C using a Reichert Ultracut E microtome fitted with an FC4D cryomicrotomy attachment. The sections were collected on formvar/carbon-coated 200-mesh copper grids and placed on drops of chilled 2% gelatin to stabilize. After three blocking steps of ammonium chloride, glycine, and goat serum, the grids were floated on drops of primary antibody for 1 hr. For both single and double labeling, the subsequent stages were essentially equivalent to those used for labeling after freeze-fracture (see above), except that instead of 5-nm and 10-nm gold-conjugated secondary antibodies 10-nm and 15-nm gold were used. After labeling and washing, the sections were stabilized in 2% methyl cellulose containing 0.4% uranyl acetate before examination in the electron microscope. The design of the control experiments was the same as that used for the fracture-label studies described above.
Confocal Laser Scanning Microscopy
For immunoconfocal microscopy, 10-μm cryosections were cut at −25C and thaw-mounted on poly-
The immunolabeled sections were examined by confocal laser scanning microscopy using a Leica TCS 4D, equipped with an argon/krypton laser and fitted with the appropriate filter blocks for detection of fluorescein and Cy3 fluorescence. Double labeled samples were imaged using simultaneous dual channel scanning. Both single optical sections and projection views from sets of 10 consecutive single optical sections taken at intervals between 0.6 and 1μm were examined. All specimens were examined within 24 hr of immunolabeling.
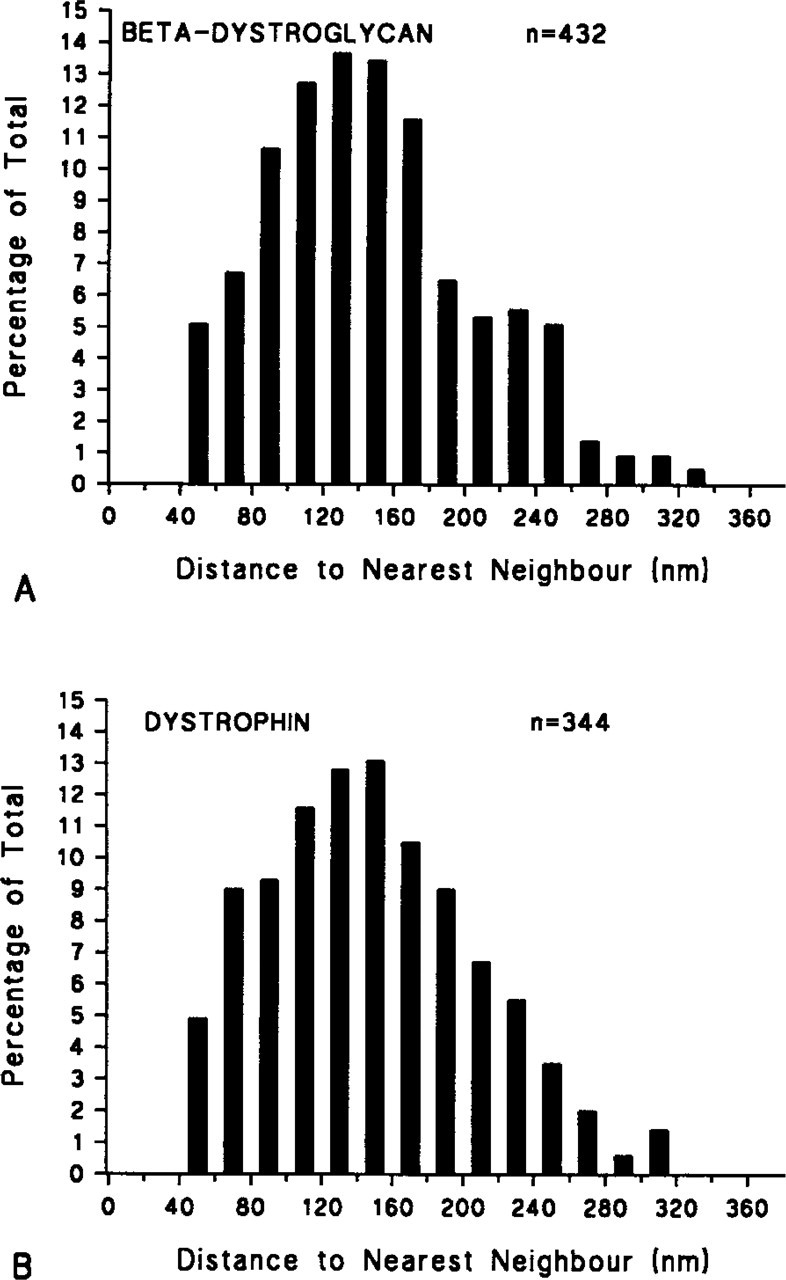
Histograms of the nearest neighbor distances of labeling sites of (
Results
Single Labeling
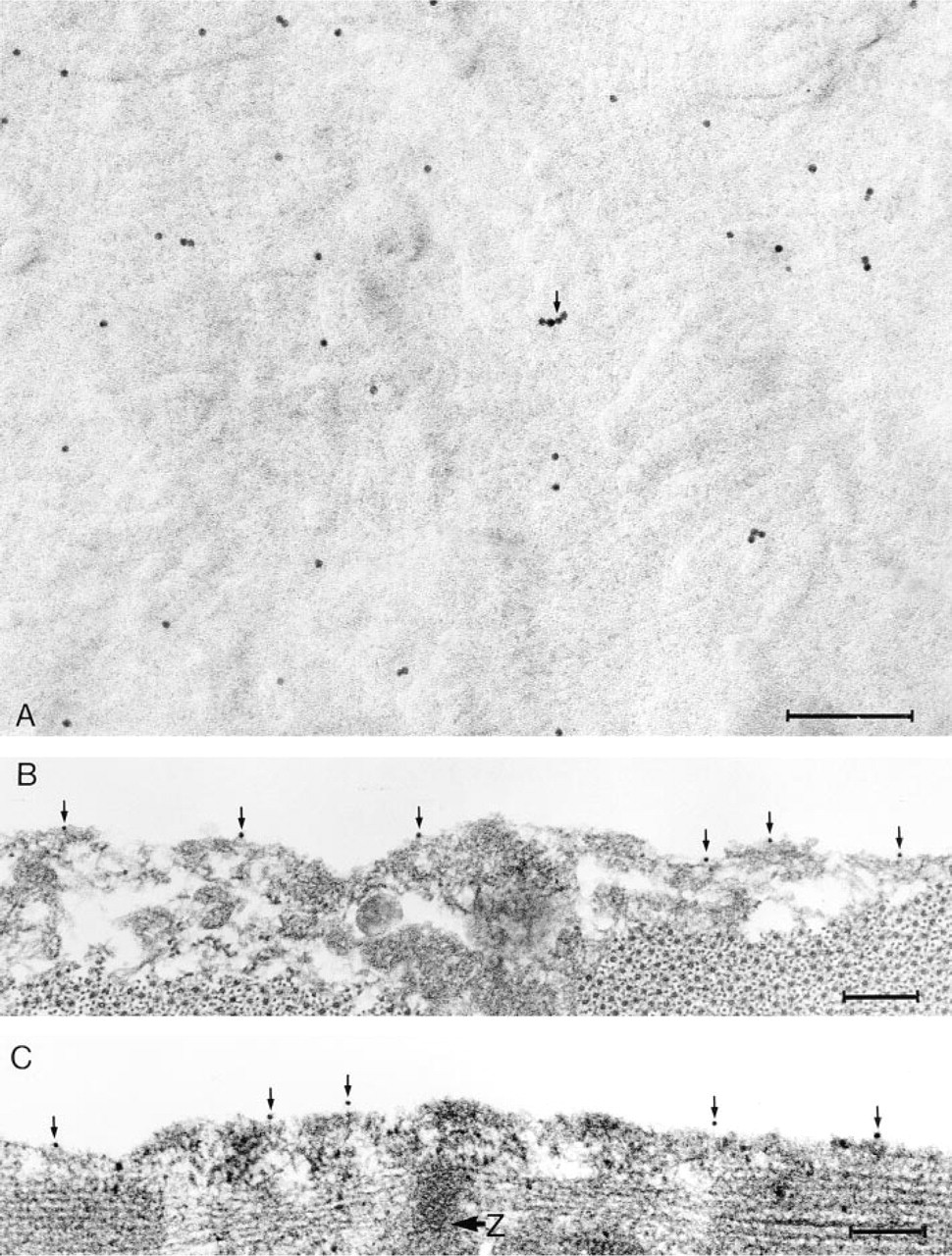
When fractured samples were labeled individually for either dystrophin or β-dystroglycan, the label was seen on those parts of the replicas that covered the P half of the plasma membrane, i.e., the half membrane leaflet left adjacent to the sarcoplasm after fracturing (Branton et al. 1975) (Figure 2A). The overall distribution of labeling sites was reasonably even, with no localized concentrations of immunogold. There were some unlabeled areas on the P half-leaflet that could be caused by loss of gold during cleaning of the replica or reorganization of the membrane during thawing (Severs 1995). Sporadic clumping of the gold particles was attributed to multiple binding to the primary antibody by the secondary conjugate (Figure 2A).
The distribution of label as seen on the platinum-carbon replicas was confirmed in sections of the resinembedded samples of the same tissue, fractured and immunolabeled in the same experiments (Figures 2B and 2C). Gold particles were positioned at the surface of the sectioned fibers, verifying that the protein molecules of the fractured surfaces were accessible for immunolabeling. In both transversely (Figure 2B) and longitudinally (Figure 2C) orientated fibers there appeared to be no preferential labeling of zones corresponding to specific segments of the underlying myofibrils.
Histograms of the nearest neighbor distances of dystrophin and β-dystroglycan labeling sites on the platinum-carbon replicas showed a very similar distribution with matching modes and means (mean ± SD 149.26 ± 59.57 and 147.24 ± 59.67, respectively) (Figure 3). The distributions of the nearest neighbor distances suggest that the positions of the sites are not completely random and that there is some regularity of structure. This was confirmed by comparing the means for the observed data with means calculated from 19 computer-simulated patterns of sites, each forming a completely random pattern and containing the same number of points as the number seen experimentally (technically a realization of a two-dimensional Poisson process). The mean nearest neighbor distances for all 19 simulated distributions were greater than the observed distances, providing strong evidence that there is an underlying systematic pattern to the positioning of the labeling sites.
Double Labeling
When fractured samples were labeled consecutively for dystrophin and β-dystroglycan, pairs of gold particles of different size could frequently be detected Figure 4). The members of each pair were often in very close proximity to each other. As with single labeling, there was occasional clumping of the gold probe, again attributed to multiple labeling of the primary antibody by the secondary conjugate. This was more common with the 5-nm particles than the 10-nm particles, because the smaller markers are less subject to steric hindrance. Some 10-nm particles showed no indication of being associated with a 5-nm particle, but if the smaller particle lay directly above or below the larger one it would not be optically detectable in the electron beam. Therefore, the percentage of sites that in fact show double labeling may be greater than is immediately apparent. The very close proximity of the gold particles in each pair, along with the low percentage of single particles, shows clearly that the pairing is not occurring by random association.

(
Close co-localization after double labeling of dystrophin and β-dystroglycan was also demonstrated when the procedure was carried out on cryosections. Gold particles of both sizes were seen in close proximity to the plasma membrane (Figure 5). Further evidence for the co-localization of the C-termini of dystrophin and β-dystroglycan was provided by confocal laser scanning microscopy through simultaneous visualisation of the fluorescent probes by dual-channel imaging (Figure 6). In the combined image, the two individual images coincide almost exactly. The overall labeling pattern was one of a continuous distribution at the fiber periphery with occasional repeating points at which it appeared to be more concentrated.
Discussion
In this study we sought to establish the spatial relationship between the C-terminal domains of dystrophin and β-dystroglycan at the plasma membrane of skeletal muscle by applying double immunogold electron microscopy with complementary dual-channel scanning immunoconfocal microscopy. To achieve this, we applied the fracture-label technique, which permits high-resolution visualization of labeled membrane components both in sections and in platinum-carbon replicas of the fractured tissue face (Pinto da Silva et al. 1986; Severs 1995). Investigations involving the double labeling of dystrophin and β-dystroglycan have been made by Wakayama's group using either mouse (Wakayama et al. 1995) or human muscle (Inoue et al. 1996). They describe 5-nm and 10-nm gold particles lying close to the plasma membrane with approximately 25% of either size of particle occurring in “doublets” with one of the alternative size. We have extended these findings by labeling freezefractured surfaces, thus obtaining unique en face views of the distribution of double labeling sites. For the purpose of this study, this approach proved particularly informative, providing novel information on the spatial relationship of concomitantly localized C-terminal domains of dystrophin and β-dystroglycan in a plane parallel with the plasma membrane surface. An important feature of the fracture-label technique is that cytochemical labeling is carried out after membranes in the sample have been split by freeze-fracture and the sample thawed. The label thus has direct access, in principle, to the entire en face aspect of suitably fractured membranes. Both integral membrane proteins and associated peripheral proteins are rendered accessible for labeling by this procedure because, on contact with aqueous media at the thawing stage, the fractured half membrane leaflets become reorganized into a discontinuous bilayer, thereby exposing associated cytoplasmic or extracellular components (Pinto da Silva et al. 1981b). This feature makes fracture-label particularly appropriate for gaining access to proteins such as dystrophin and the cytoplasmic domain of β-dystroglycan, which lie just beneath the inner surface of the plasma membrane.
In previous double labeling studies we have examined proteins that, by fluorescence imaging, may appear to co-localize but which, at the resolution of the electron microscope, can be identified as being situated in different cellular compartments (Cullen et al. 1996; Stevenson et al. 1997). Dystrophin and merosin, for example, appear to co-localize closely by immunofluorescence imaging but, in fact, by immunogold labeling can be seen to lie on opposite sides of the plasma membrane (Cullen et al. 1996). In the present study, by contrast, we have compared the localization of two proteins using antibodies to their C-termini, which by all criteria appear to co-localize exactly. Labeled individually at the fractured membrane surface, images of the two proteins are indistinguishable in terms of site distribution and nearest neighbor distance. When double labeled, the gold particles in each pair are usually closely adjacent and frequently in contact with each other. In theory, after double labeling, two gold particles labeling two adjacent epitopes can, in the worst case, be separated by the length of four antibody molecules, i.e., approximately 40 nm (Harris and Cullen 1992). In the present study, the gold particles in each pair were in the great majority of cases separated by considerably less than 40 nm and can therefore be considered to be truly co-localizing.
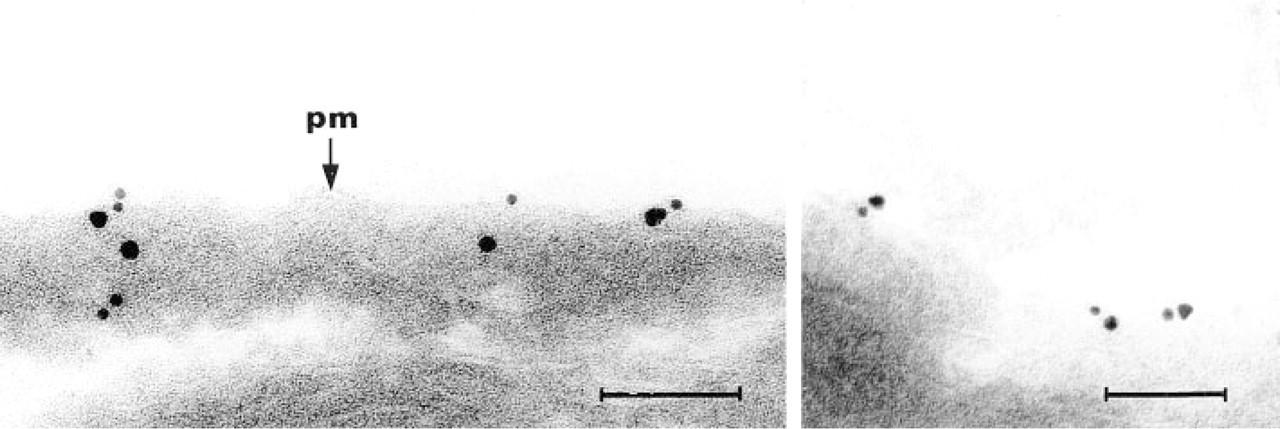
Two examples of double immunogold labeling of dystrophin (10-nm gold) and β-dystroglycan (15-nm gold) carried out on cryosectioned rat skeletal muscle. Gold particles are seen in close proximity to the plasma membrane (pm) and to each other. Bars = 100 nm.
The interest in the confocal microscopy is not so much that it shows the expected co-localization but that it provides a broader scan of the overall distribution of the proteins. Earlier immunofluorescence work has suggested that dystrophin is arranged in costameres, i.e., that it forms a band at the level of the Z-line or either side of the Z-line (Masuda et al. 1992; Minetti et al. 1992; Porter et al. 1992; Straub et al. 1992). This costameric distribution has not been supported by ultrastructural evidence. From our own observations, there are occasional increases in the intensity of fluorescence with a repeat corresponding to the length of a sarcomere, but the distribution of both dystrophin and β-dystroglycan is nevertheless continuous over the whole of the myofiber sarcolemma. This is similar to our observations on cardiac muscle in which, however, the components of the DGC also penetrate the tubules of the T-system (Stevenson et al. 1997). Our observations of gold particle distribution on the freezefracture replicas also do not provide evidence of a costameric association. This is in accord with our findings from studies using cryosectioned material (Cullen et al. 1990,1991,1994; Cullen and Watkins 1993). Interestingly, in relation to our own results, Wakayama's group has also found that their attempts to identify costameric structures were “not consistent.”
In conclusion, we have shown that the C-termini of dystrophin and β-dystroglycan are extremely closely associated at the cytoplasmic surface of the plasma membrane. The separation between the two epitopes is on the same order as the size of the gold probes (5-10 nm) or less than this. This is consistent with the biochemical evidence that dystrophin is bound to β-dystroglycan and is of interest because, whereas the binding site on β-dystroglycan is known to be at the C-terminus (Jung et al. 1995; Rosa et al. 1996), on dystrophin it is on the cysteine-rich domain and the first half of the C-terminal domain (Suzuki et al. 1994). Recently it has been shown that the second half of the C-terminal domain, more particularly those amino acid residues encoded by exons 73-74, bind α1- and β1-syntrophin, which are cytoplasmic proteins of the glycoprotein complex (Figure 1) (Ahn and Kunkel 1993; Suzuki et al. 1994). The epitope we labeled in the present study, the last 16 amino acids encoded by exon 79, lies beyond the syntrophin binding site. Therefore, our results suggest that the tip of the dystrophin molecule folds back towards the dystroglycan binding site or that the syntrophins are of a shape that does not prevent near apposition of the tips of the molecules of dystrophin and β-dystroglycan. We intend to examine these interrelationships further by extending our immunolabeling studies to include more of the components of the DGC, in particular the syntrophins.

Simultaneous confocal visualisation of dystrophin and β-dystroglycan by dual-channel imaging of double labeled sections. Immunolabeling for dystrophin (green fluorescence) and β-dystroglycan (red fluorescence) is shown independently in panels
Footnotes
Acknowledgements
MJC and JW were supported by The Wellcome Trust, grant number 046045/Z/95/Z; SS, SR, and NJS were supported by The British Heart Foundation, grant numbers FS/94044 and PG/93136.
We thank Dr Henry Klamut (Ontario Cancer Institute, Toronto) for the gift of the polyclonal antibody P1583, Dr Louise Anderson (Dept of Neurobiology, University of Newcastle upon Tyne) for the monoclonal antibody 43DAG/8D5, and Dr Trevor Cox (Dept of Mathematics and Statistics, University of Newcastle upon Tyne) for assistance with the spatial statistics.