
Abstract
The application of immunoelectronmicroscopy to soluble proteins is limited because soluble proteins can redistribute during fixation. Fixation may also adversely affect the recognition of proteins associated with membranes. We show here how displacements of soluble proteins can be prevented and antigen sensitivity improved by freeze-substitution immunocytochemistry. The usefulness of this method for soluble cytoplasmic proteins is demonstrated for the twitchin protein in Aplysia muscle and the kinesin motor proteins in squid giant axons, in which the sizes of various cytoplasmic pools of kinesins are estimated. The utility for membrane proteins present in small numbers of copies is demonstrated by labeling a glutamate receptor subunit in mouse cerebellar cortex and the ZO-1 protein in tight junctions between MDCK cells. Thus, freeze-substitution immunocytochemistry can show the native distribution of both soluble and membrane proteins labeled with polyclonal antibodies and, at the same time, can reveal structural features comparable to those in chemically fixed or osmium freeze-substituted samples.
C
There are several reasons to doubt that current immunochemical methods give an accurate picture of the distribution of soluble or readily solubilized proteins in a living cell (Melan and Sluder 1992). Traditional methods involve the application of chemical crosslinkers that must be presented to the cell in a buffer solution of unknown compatibility with the proteins and their binding properties. Moreover, a crosslinker binding to a soluble protein would be expected to bind to any fixed protein, such as components of the cytoskeleton, that might lie within diffusing distance. Therefore, application of a fixative could release loosely bound proteins, making them soluble, and then link them to other cytoplasmic elements with which they did not originally associate (Orkand and Kravitz 1971). The use of chemical fixatives on some membrane proteins could even alter the actual structural relationship of the protein with the membrane. In addition, configurational changes or protein denaturation could render an epitope inaccessible to an antibody.
A solution to these problems might start with a physical fixation, such as rapid freezing, which would have the advantages that no fixatives would have to diffuse into the cell and that the fixation could be completed in a fraction of a msec (Heuser et al. 1979; Bridgman and Dailey 1989; Monaghan and Robertson 1990) Once frozen, the problem becomes how to make individual soluble antigens available to the antibody without displacing the antigens. The difficulty is that the antibody must reach the antigen by diffusion but the antigen cannot be allowed to diffuse.
A solution to this conumdrum is to use a freeze-substitution method (Bridgman and Dailey 1989) to stabilize the tissue at low temperatures in a nonaqueous environment, in which diffusional movement would be limited to substances soluble in cold acetone. Proteins are then held in place by low-temperature embedding in a Lowicryl resin, leaving some antigenic sites exposed on or near the surfaces of sections. Inclusion of uranyl acetate during freeze-substitution stabilizes and stains membranes, resulting in retention of structures comparable to those seen after conventional preparation for thin sectioning.
We expected this method to be less sensitive than the pre-embedding methods but we also expected that much of the redistribution of proteins during processing for immunocytochemistry would be prevented. We were pleased to find that low background staining compensated for the decreased sensitivity. Although our initial interest was in the overall distribution of the kinesins, this method appeared to have wider potential applications when we tested it with several other preparations that ordinarily present difficulties with localizing soluble proteins or membrane-associated proteins present in small amounts. Here we present the results with localizing the kinesins in the squid giant axon as well as the results with other soluble and membrane-associated antigens.
Materials and Methods
All tissues were first rapid-frozen with a Life Cell CF-100 freeze-slamming apparatus (Life Cell; The Woodlands, TX) (manuscript in preparation). Squid (Loligo pealii) axons were extruded onto a square of 4% agar lying on an aluminum freezing stage (a disk of aluminum 13 mm in diameter to which a circle of filter paper, 6 mm in diameter, is attached with epoxy glue) (Heuser et al. 1979), so that a column of axoplasm could be frozen along its longitudinal axis. Samples of the accessory radula muscle of Aplysia californica were mounted on the freezing stage with its muscle fibers parallel to the freezing block. MDCK cells (American Type Culture Collection; Rockville, MD), were cultured on Thermanox coverslips at 37C in air/5% CO2 (Taub et al. 1979). The coverslips were also mounted on squares of agar on the freezing stage. A lateral lobe of the cerebellum was removed from 25-30-day-old C57-B1/6 mice and laid on the freezing stage with the pial surface facing up before freezing.
Kinesin was labeled with an affinity-purified rabbit IgG made to a 394 amino-acid synthetic peptide from the N terminus of the motor domain of squid kinesin heavy chain (SK-394) (Muresan et al. 1996). A polyclonal antibody against the N-terminal fragment of squid neurofilament recognizes the three neurofilament proteins at 60, 70, and 200 kD (N-terminal) (Grant et al. 1995). SK-394 recognized a double band at 116 kD in immunoblots of homogenized squid axoplasm as well as in the pellet and supernatant (in 500 mM NaCl with protease inhibitors; centrifugation at 130,000 × g for 10 min). The anti-squid neurofilament antibody recognized bands at 60, 70, and 200 kD in blots prepared with the same protocol (Figure 1B). The polyclonal antibody against ZO-1 was obtained from Zymed (San Francisco, CA). A polyclonal antibody against a glutamate receptor α-subunit was a gift from Ronald Petralia (Mayat et al. 1995), and against Aplysia twitchin from Ferdinand Vilim (Probst et al. 1994).
Frozen samples for immunocytochemistry that were freeze-substituted on their specimen carriers were rocked at −80C in 0.1% uranyl acetate in acetone for 24 hr, warmed up to −60C, and rinsed twice in acetone (10 min each) and once in methanol. Freshly mixed K11M resin was bubbled at room temperature (RT) with dry nitrogen to mix components and to reduce contact with oxygen. Infiltration with resin diluted to 50% in acetone was followed by 75% resin and then by two changes of 100% resin at −60C, allowing 24 hr for each change. Samples were then placed in aluminum dishes, sealed with Saran plastic wrap, and polymerized with 300 nm UV for 2 days, starting at −60C, and warming up in 12-hr steps to −40C, −10C, and RT. After polymerization, the samples were placed under vacuum overnight. Cryopolymerization by UV takes almost 48 hr to complete, so there is little possibility of heating from the exothermic reaction (Glauert and Young 1989).
Samples of axoplasm for structural comparison were prepared by substituting them at −80C with osmium tetroxide in acetone containing 4% osmium tetroxide for 36 hr (Moreira et al. 1996). Samples were then passively warmed to −20C, transferred to crushed ice for 1 hr, block-stained with 1% uranyl acetate in acetone, rinsed briefly in methanol, substituted with propylene oxide, and embedded in Araldite resin (CY212).
All immunocytochemistry was performed on conventional thin plastic sections. These were cut at 70-90 nm on water in a Reichert Ultracut S ultramicrotome, collected on Formvar-coated nickel grids, and immunostained with protein A-gold without etching (Moreira et al. 1991). Grids were sequentially floated on 1% BSA in 0.02 M Tris-HCl, 0.5 M NaCl (TBS, 30 min), primary antibody diluted 1:10-1:50 in TBS (overnight, 4C), and protein A-gold (15 nm; BioCell Research Laboratories, Cardiff, UK) diluted 1:20 in TBS (1 hr). Rinses with TBS were done after each step and finally in distilled water before drying and staining with uranyl acetate and lead citrate. Controls included incubation of grids following the same procedures but omitting the primary antibody, incubation with a nonimmune rabbit IgG as a primary antibody, or incubation with the preimmune IgG diluted in TBS.
Quantitative analysis was performed with a Zidas digitizing system (Carl Zeiss; Thornwood, NY) interfaced with a Macintosh computer. Sets of five grids of three different axoplasm preparations were incubated with the antibodies SK 394, neurofilament, or their corresponding preimmune sera. Thirty micrographs from each condition—kinesin, neurofilament, and control—were taken from the edge where the tissue had contacted the freezing block.
The cytosolic area was measured by subtracting the area corresponding to each organelle from the total area of the photograph in micrographs at × 50,000 final magnification. Conventions for counting were based on the dimensions of kinesin because vesicles moving along microtubules lie 16-18 nm from the microtubule (Miller and Lasek 1985; Hirokawa et al. 1989). Thus, gold particles lying up to 42 nm (i.e., 17 nm, plus the combined lengths of the IgG and the protein A, ~ 25 nm) inside or outside of the organelle limiting membrane were considered to be tagging an attached kinesin molecule. Gold particles more than 42 nm from the organelle were considered to label kinesin in the cytosol. In the rare instances when a gold particle lay in the intersection of the domains belonging to two different organelles, it was arbitrarily assigned to the organelle with the lesser amount of gold.
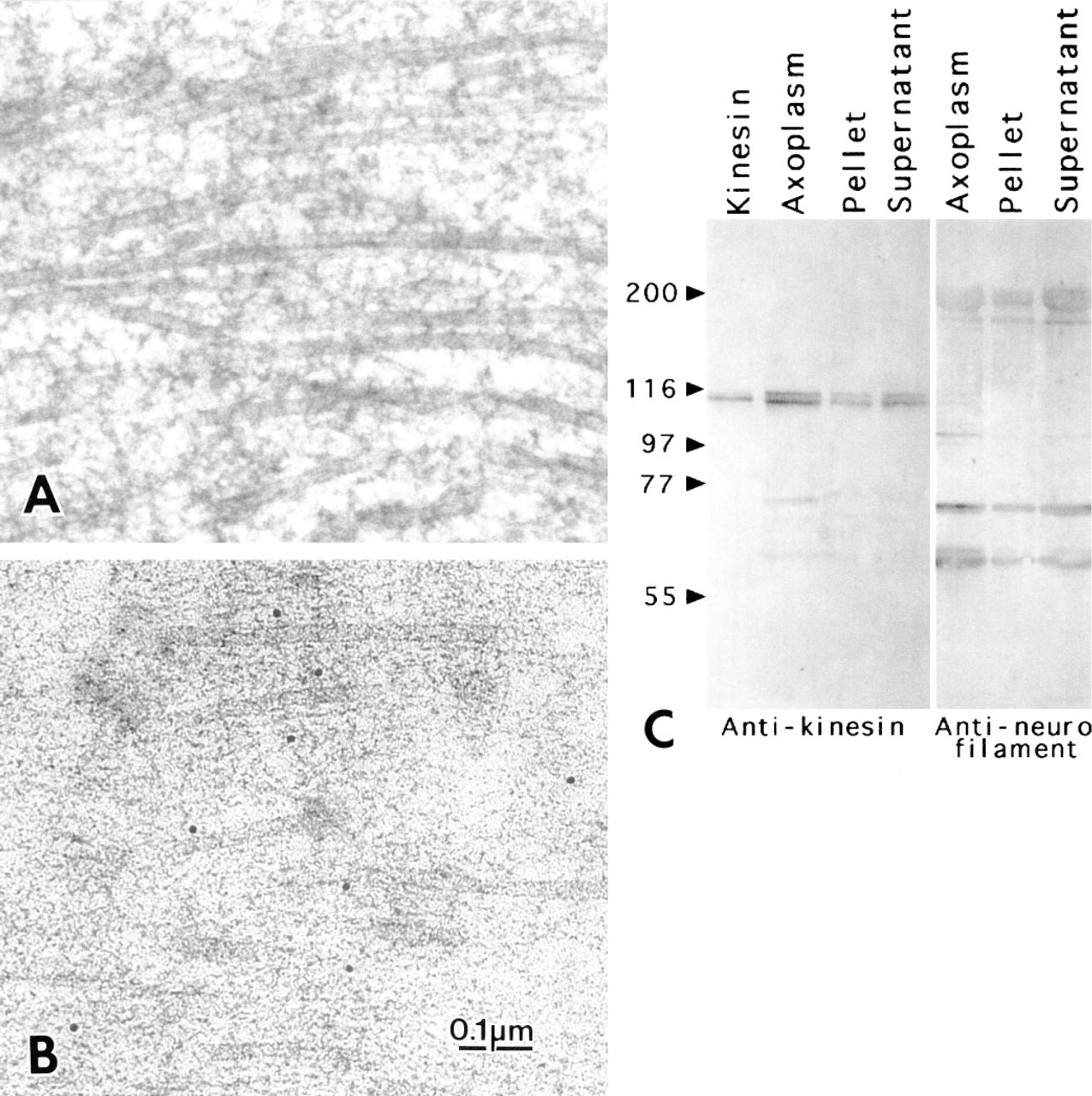
Longitudinal thin sections of extruded squid axoplasm.
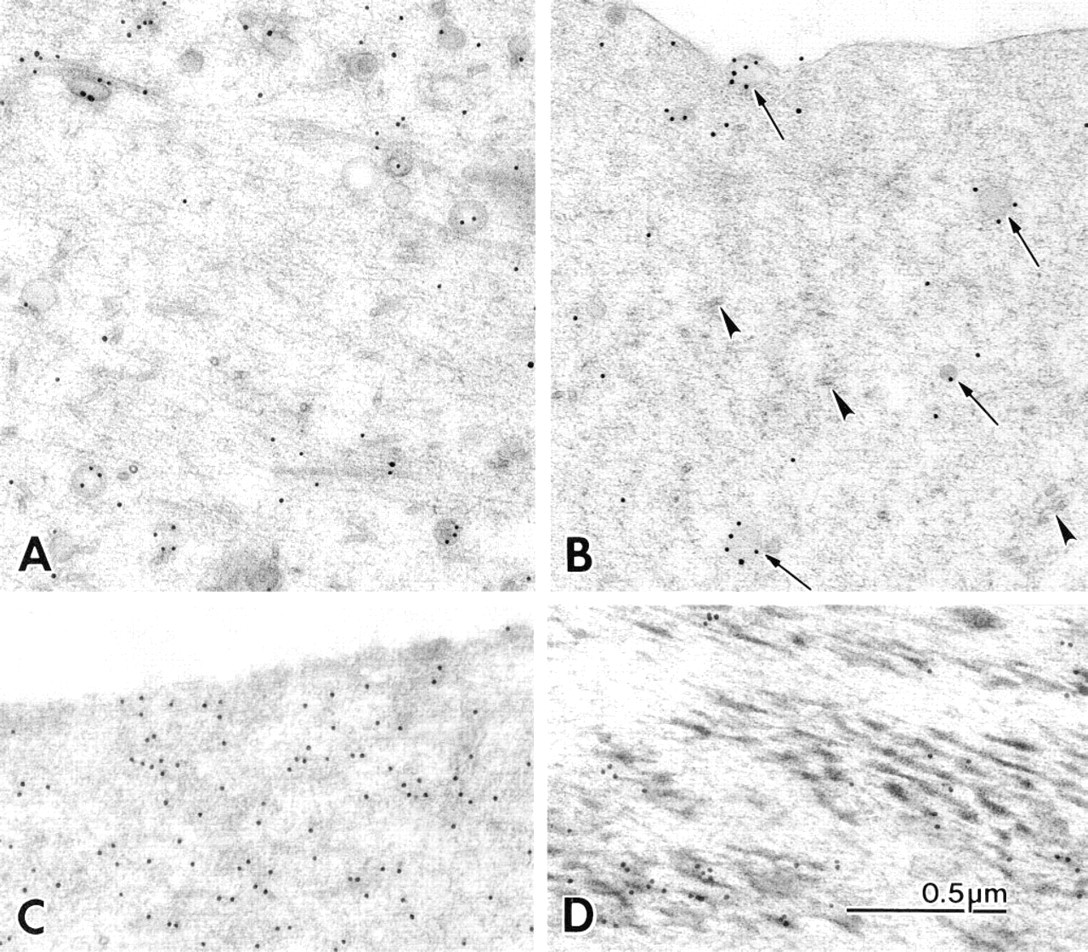
(A,B) Longitudinal thin sections of freeze-substituted squid axoplasm immunostained for kinesin.
Results
Axoplasm
Axoplasm prepared by conventional freeze-substitution (Moreira et al. 1996) could be examined up to 10-15 μm from the freezing surface, where it disappeared into ice crystal patterns. Axoplasm substituted with osmium was similar to that in axons fixed by immersion in glutaraldehyde and postfixed with osmium (see Miller and Lasek 1985) in showing a thick mat of indistinct cytoskeletal elements, among which only microtubules and membranes were clearly differentiated (Figure 1A).
Samples substituted for immunocytochemistry in the absence of osmium showed finer and more distinct cytoskeletal details than those substituted with osmium (Figure 1B). Among the various longitudinal cytoskeletal elements, the parallel microfilaments are presumed to be principally neurofilaments (Cohen et al. 1987) and actin filaments (Bearer et al. 1993). Microtubules appeared as distinct tubules running approximately parallel to the microfilaments. Densecored and clear vesicles of various diameters, ER cisternae, and mitochondria were also apparent. Cross and tangential views of microtubules occasionally seen in the longitudinal sections are assumed to represent rearrangements occurring during extrusion (Figures 2A and 2B). In any instance, axoplasmic structures were shown in good structural detail in axons prepared for immunocytochemistry.
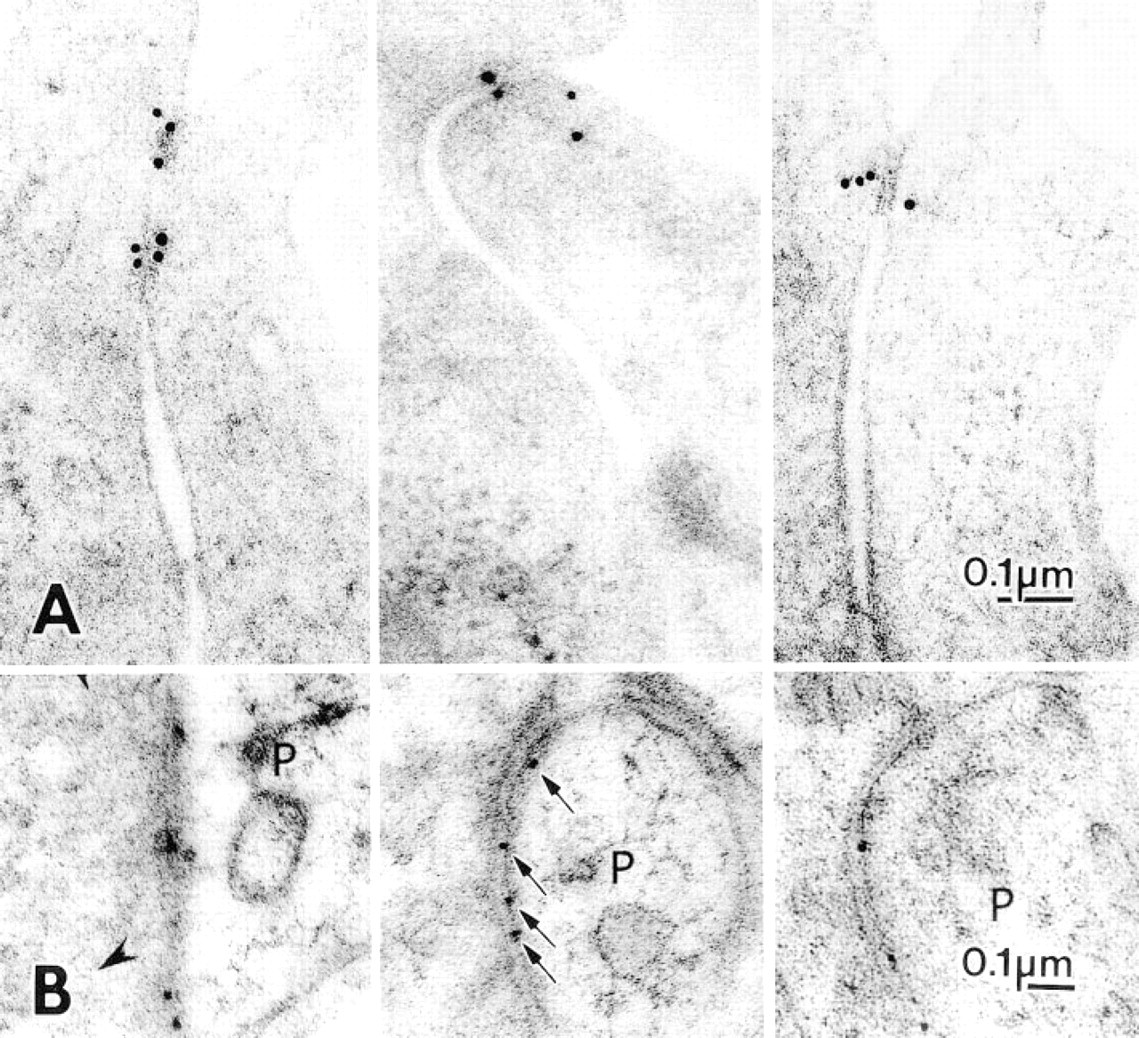
(A) Gold particles (15-nm) mark ZO-1 proteins in cultured MDCK cells prepared by freeze-substitution. Despite the lack of osmium in the substitution medium, the limiting plasma membranes at the tight junctions and their relationships with the adjacent gold particles are clearly visualized.
Structure of Muscle, Cerebellum and Cultured Cells
Aplysia muscle and MDCK cells substituted without osmium and embedded in Lowicryl showed well-defined cytoplasmic structure and membrane outlining. Tight junctions on the MDCK cells and their anchoring filaments showed a clarity of structure comparable to similar structures reported previously with aldehyde fixation (Figure 5 in Stevenson et al. 1989). Synapses in cerebellar cortex typically lay somewhat beyond the 15-μm limit of optimal structural preservation, but images of pre- and postsynaptic terminals still permitted delineation of the distribution of gold label along synaptic densities (Figure 3A).
Immunoelectron Microscopy
Various combinations of aldehyde fixatives, as well as several different embedding resins, were tried in conjunction with freeze-substitution, but substantial labeling was obtained only with Lowicryl K11M low-temperature embedding after substitution without fixatives. The kinesin antibody followed by protein A-gold (15 nm) provided a large amount of labeling with little nonspecific background.
The SK-394 labeling in the axoplasm showed gold particles more concentrated around organelles and microtubules, although organelles varied considerably with respect to the amount of labeling on their surfaces. The most consistently labeled organelles were vesicles, which could have up to five gold particles uniformly distributed around their perimeter. Gold particles were distributed rather than clustered at vesicle surfaces, suggesting that neither the kinesins on the vesicle surface nor the protein A-gold complexes had clumped (Figures 2A and 2B).
Labeling with the neurofilament antibody was performed in parallel with kinesin labeling to compare the distribution of a widely distributed and relatively stable cytoskeletal element. Neurofilament label appeared as a faintly linear arrangement of gold particles throughout the axoplasm, without any increase around organelles or microtubules (Figure 2C). The twitchin protein is another example of a soluble protein that could be localized in Aplysia muscle tissue by the freeze-substitution method. Gold particles were clearly associated with the contractile elements, often concentrated at their ends (Figure 2D).
The antibodies against membrane proteins—ZO-1 and a glutamate receptor—produced precise localizations with almost no background. After incubations with anti ZO-1 protein, discrete distributions of gold particles showed a distribution at the edges of tight junctions consistent with the expected location of the ZO-1 protein (Figure 3A). The δ-subunit of a glutamate receptor present in cerebellar cortex synapses was marked by three to five gold particles regularly distributed along postsynaptic densities in Purkinje cell dendritic spines (Figure 3B), and therefore was also distributed exactly as expected.
Measurement of the Distribution of Kinesin Label
The localizations of ZO-1 and glutamate receptor were so focal and accompanied by so little background that they did not require morphometric analysis. The kinesins were, however, expected to be distributed throughout several cellular compartments, including the cytoplasm. The distribution of gold particles corresponding to SK-394 anti-kinesin, as well as anti-neurofilament labeling, was measured particle by particle on photographic prints comparable to those in Figure 2. Vesicles accounted for 36.4% and ER for 8.9% of the total gold count, whereas mitochondria were only 1.3% of the total (Table 1). Added together, the organelles accounted for almost half of the total cytoplasmic label. The other half was distributed between microtubules (31.4%) and the cytosol (21.9%). Because the background label over empty plastic was 0.3 particles/μm2, the contribution of background to the distribution of organelle label was negligible, although it could account for as much as 15% of the raw cytoplasmic label. Whereas half of the kinesin labeling was in cytosol and associated with microtubules, label was concentrated 57-fold around vesicles compared to cytosol, 23.5-fold around ER cisternae, 12-fold around microtubules, and fivefold around mitochondria (Table 1).
Discussion
The labeling method used here combines structural definition sufficient to recognize individual cytoskeletal elements and organelles with sensitive labeling of cytoplasmic proteins. The structure of axoplasm prepared by this method is, in general, comparable to that of axoplasm substituted with osmium tetroxide. However, the finest cytoskeletal elements, which are typically obliterated or coagulated by the osmium substitution, are clearly visible in the axoplasm prepared for immunochemistry.
The accelerated substitution protocol was designed to provide minimal opportunity for soluble cytoplasmic proteins to diffuse during specimen preparation (Bridgman and Dailey 1989) . Indeed, the results with squid axoplasm and Aplysia muscle suggest that this goal was achieved, as would be expected on consideration of the protocol. The surface of the axoplasm is stabilized in less than a msec by contact with the freezing block (Heuser et al. 1979) and is then substituted in acetone at −80C. The substitution step is designed to remove the ice but leave behind proteins to associate with the nearest cytoskeletal structure, which would require movements amounting to no more than a few nanometers. The infiltration and polymerization in acrylic resin are done at −60C, conditions in which proteins would be insoluble. At worst, individual proteins might move far enough to rest against immediately adjacent cytoskeletal or other elements of the cytoplasmic matrix. In contrast, proteins can move micrometer distances during aldehyde fixation (Orkand and Kravitz 1971).
Distribution of immunogold in squid axoplasm
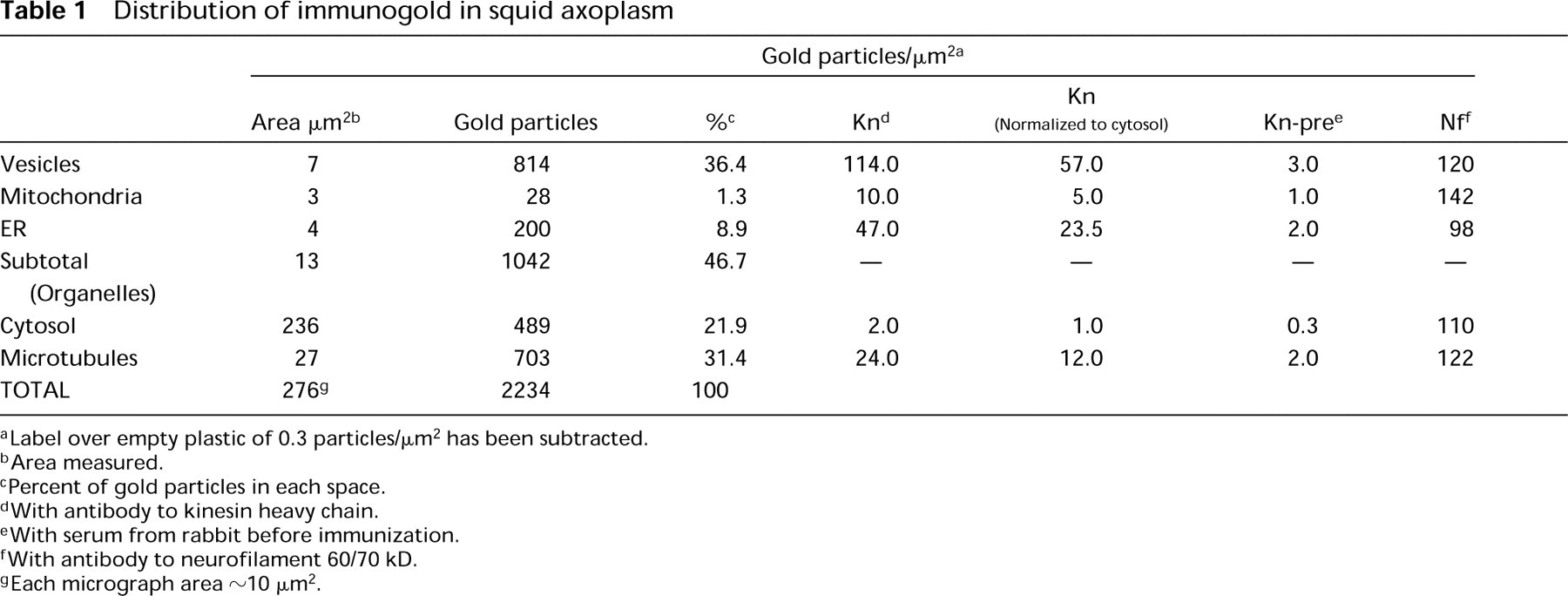
Label over empty plastic of 0.3 particles/μm2 has been subtracted.
Area measured.
Percent of gold particles in each space.
With antibody to kinesin heavy chain.
With serum from rabbit before immunization.
With antibody to neurofilament 60/70 kD.
Each micrograph area ~ 10 μm2.
Another advantage of the freeze-substitution method is that it appears to enhance the sensitivity of the antibody labeling in the absence of a crosslinking fixative. Furthermore, the background labeling, to judge by the amount of label over regions lacking tissue, is very low (see Table 1), resulting in a very high signal-to-noise ratio. Thus, an antibody that recognizes many members of the kinesin family, such as AK-493, permitted the relative partitioning of various kinesins between different cellular compartments to be estimated from the distribution of gold grains.
The present results are in agreement with current biochemical studies of kinesin (Vale et al. 1985) but differ somewhat from previous studies with conventional immunocytochemistry, in which much higher percentages of organelle labeling against higher backgrounds are reported (Hirokawa et al. 1991). The present results confirm that there is a significant pool of kinesin in axoplasm that is not associated with membranous organelles. Although the concentration of these kinesins is low relative to organelles, the amount calculated from the distribution of gold grains is about one half of the total kinesin when the much larger volume of the soluble compartment is taken into account. Migration of the soluble kinesin in crosslinking fixatives, a decreased sensitivity of labeling in the absence of fixation, and extraction of the free kinesin in the aqueous fixative can account for previous failure to detect the soluble pool of kinesin by immunofluorescence and cytofluorimetric study (Dahlstrom et al. 1991), or by immunoelectron microscopy of ultrathin frozen sections (Hirokawa et al. 1991). Cryosectioning for immunolocalization of proteins provides sufficient sensitivity but is not suitable for soluble antigens because the tissue must still be lightly fixed with aldehydes.
The substitution method appeared to detect kinesin attached to axoplasmic organelles as well as the soluble kinesins. The separations of gold particles clustered around organelles from the organelle surfaces were consistent with what is known about the lengths of kinesin bridges, when the size of the IgG-protein A complex is accounted for (Miller and Lasek 1985; Hirokawa et al. 1989). In addition, the gold label on mitochondria is localized in small clusters, consistent with the interpretation of video images of mitochondria moving along microtubules, which suggests, that kinesin has a patchy distribution on mitochondrial surfaces (Nangaku et al. 1994).
Another soluble protein, twitchin, that may participate in excitation-contraction coupling is abundant in muscle fibers of invertebrates, but its localization in Aplysia had proved difficult to determine by conventional immunocytochemical methods (Probst et al. 1994). However, twitchin in Aplysia was localized in the expected distribution at the ends of thick filaments by the present method, whereas methods using fixatives, such as low concentrations of glutaraldehyde or freeze-substitution with osmium tetroxide, showed sparse gold particles distributed over the empty plastic as well as the muscle (F. Vilim, personal communication; Probst et al. 1994).
Different types of organelles would be expected to have different species of kinesin on them (Hirokawa 1998), but the AK-493 polyclonal antibody was designed to recognize many of them and thus to show the overall distribution of many of the kinesin types. Attempts to separate kinesins from organelles isolated from squid axoplasm have showed that the kinesins which move them are, in fact, tightly bound to their surfaces (Schnapp et al. 1992). Because immunocytochemistry does not distinguish between tight binding and loose associations, it remains difficult to assign a functional significance to the pool of kinesin found on microtubules and ER. Kinesin does have a second binding site for microtubules outside of its motor domain (Andrews et al. 1993), and ER isolated from squid axoplasm is known to move along micro-tubules (Dabora and Sheetz 1988).
Proteins associated with tight junctions, such as ZO-1, have been localized with immunoelectron microscopy using pre- or postembedding procedures with only dilute paraformaldehyde as a fixative (Stevenson et al. 1989; Itoh et al. 1993). These approaches yield valuable information on the in situ localization of the proteins, but the loose clustering of gold particles around the tight junctions might mean that details of the distributions of labeled proteins at the junction were not maintained. Using the present method, localization of the ZO-1 protein with tight alignment of the gold particles near the junction appears to depict more precisely the arrangements of ZO-1 proteins at tight junctions in MDCK cells. Background labeling elsewhere in the cells was very low, so most of the labeling at the tight junction can be taken as representative of the distribution of ZO-1 protein.
For proteins lying in the plane of a membrane, such as metabotropic glutamate receptors, localization has been possible using pre-embedding methods and chemical fixation (Shigemoto et al. 1996). Small (1-nm) gold particles conjugated to the secondary antibody had to be used to gain entry to the synaptic cleft and then were silver-enhanced, leaving silver particles that covered much of the postsynaptic density. With the present freeze-substitution method, an α-subunit of a glutamate receptor was clearly decorated by an antibody followed by 10-nm protein A-gold, showing specific postsynaptic localization. In this case, the distribution of gold particles just large enough to detect against the synaptic cleft and postsynaptic membrane material provides a precise localization of the receptor along the postsynaptic membrane at synapses of parallel fibers on Purkinje spines (Zhao et al. 1997).
The addition of uranyl acetate with unfixed samples does not reduce labeling (Humbel and Schwarz 1989) but greatly enhances membrane preservation and yields good images of the cytoskeletal elements and the overall cytosolic background. Thus, the relatively simple immunolabeling protocol presented here combines sensitivity, low background, and stabilization and preservation of the distributions of soluble proteins with structural detail at membrane and cytoskeletal levels that is comparable and, in some circumstances, better than using fixation protocols.
Footnotes
Acknowledgements
We thank Dr Audrey Glauert for critical reading of the initial version of this manuscript, Mr James Tomlin for the interfacing of the Zidas digitizing system to the Macintosh computer, Dr Ayse Dosemesci for the immunoblot, Dr Harish Pant for the anti-neurofilament antibody, Dr Ronald Petralia for the incubations of the Lowicryl thin sections of cerebellum, Dr Ferdinand Vilim for the anti-twitchin antibody, and Mr John Chludzinski for expert photographic help.