
Abstract
We have previously obtained indirect evidence that sarcoplasmic reticulum (SR) vesicles from cardiac and skeletal muscle contain the complete chain of glycolytic enzymes from aldolase to pyruvate kinase. To investigate directly whether pyruvate kinase and other glycolytic enzymes are anatomically associated with the SR, electron microscopic immunogold labeling studies were carried out in isolated SR vesicles using specific primary antibodies against selected glycolytic enzymes and Ca2+-ATPase, and appropriate secondary antibodies labeled with 6-nm or 12-nm gold particles. Pyruvate kinase was broadly dispersed on the cytoplasmic side of the SR membrane of both cardiac and skeletal muscle vesicles. With 6-nm gold particles, the density of binding to pyruvate kinase was 2522 ± 445 and 4171 ± 1379 particles/μm2 for cardiac and skeletal muscle SR, respectively. Binding densities to Ca2+-ATPase were similar (2550 ± 639 particles/μm2 for cardiac SR, 3877 ± 408 particles/μm2 for skeletal muscle SR). Immunogold labeling of ultrathin sections indicated that pyruvate kinase was attached to the SR membrane and located immediately adjacent to the Ca2+-ATPase. Aldolase and glyceraldehyde phosphate dehydrogenase were also found to be attached to the cytoplasmic side of SR vesicles and located in close proximity to Ca2+-ATPase. These results provide the first ultrastructural evidence that glycolytic enzymes are anatomically associated with SR membranes and located near the SR Ca2+-ATPase. The results further support the hypothesis that ATP generated by SR-associated glycolytic enzymes is coupled to SR Ca2+ active transport.
D
On the basis of these studies, we hypothesized that SR-associated glycolytic enzymes may be structurally co-localized with SR Ca2+-ATPase, serving as a basis for their functional coupling to SR calcium active transport. Immunoelectron microscopy was chosen as the technique for this study because it offers the highest resolution currently available for specific immunogold labeling of subcellular components at the molecular level.
Materials and Methods
Materials
Monoclonal mouse anti-SERCA1 (specifically directed against skeletal muscle SR Ca2+-ATPase), anti-SERCA2 (specifically directed against cardiac SR Ca2+-ATPase), and polycolonal anti-Na+,K+-ATPase were purchased from Affinity Bio-Reagents (Golden, CO). Monoclonal mouse anti-glyceraldehyde phosphate dehydrogenase (GAPDH) and polyclonal goat anti-pyruvate kinase (PK) and anti-aldolase were purchased from Biogenesis (Sandown, NH). All of the antibodies have been carefully characterized for high antigen affinity and specificity. All of the EM grade secondary antibodies (donkey anti-mouse IgG and anti-goat IgG) were purchased complexed to colloidal gold from Jackson ImmunoResearch Laboratories (Avondale, PA). The copper grids (400 mesh) and nickel grids (200 mesh) were from Pelco (Redding, CA). All other chemical reagents were purchased from Sigma Chemical (St Louis, MO).
Isolation of SR Vesicles
Cardiac and skeletal muscle SR vesicles (right side out) were prepared from hearts and hind legs of New Zealand White rabbits as described previously (Xu et al. 1995). Briefly, the initial homogenization was carried out twice in 0.29 M sucrose and 10 mM imidazole-HCl buffer without KCl, pH 6.8, for 15 seconds at 22,000 rpm. The homogenate was filtered through six to eight layers of cheesecloth, centrifuged using a JA-10 rotor at 8000 rpm for 15 min, and the pellet discarded. The supernatant was filtered again with six to eight layers of cheesecloth and recentrifuged using a Ti 45 rotor at 30,000 rpm for 90 min. The pellet was harvested, resuspended, and placed on a sucrose step gradient (45, 38, 34, 32, 26, and 20%). Centrifugation was carried out using a SW-28 rotor for 16 hr at 20,000 rpm. The SR fraction at the interfaces between the 32 and 34% gradient steps was collected and sedimented for 90 min at 34,000 rpm. The final pellet was resuspended in 10 mM imidazole-HCl, 0.29 M sucrose buffer, pH 7.2, and stored at -70°C.
Immunoelectron Microscopy of Whole-mount SR Vesicles
Copper mesh grids were freshly coated with a 50-nm thin layer of colloidon (from 1% parlodion in amyl acetate), stabilized with carbon in a Polaron evaporator, and glow-discharged for 15 sec before use. SR vesicles were diluted 1:200 in 0.01 M PBS (NaH2PO4/Na2HPO4, NaCl 0.14 M, pH 7.2) and adsorbed to the grids for 2 min. All subsequent rinses and incubations were done by grid flotation on 50-μl drops of solution at room temperature. All solutions were filtered with a 0.22-μm Gelman filter except for antibody solutions. Grids were transferred with a #5 miracle-tip electron microscopy (EM) forceps, which was cleaned with distilled water and wiped dry with lint-free lens paper between each step.
After vesicle adsorption, grids were rinsed in PBS for 1 min, then blocked in 5% horse serum for 10 min. After three 1-min rinsings in PBS, samples were incubated with the appropriate primary antibody diluted 1:200 in PBS for 30 min. The grids were then rinsed with PBS to remove excess unbound antibody molecules and incubated with donkey anti-mouse or anti-goat IgG conjugated to 6-nm colloidal gold, diluted 1:40 in PBS for 30 min. Controls included preparations without the primary antibody or with substitution of an antibody against the β-subunit of the Na+,K+-ATPase for the specific primary antibody. At the end of the incubation the grids were thoroughly washed with PBS and distilled water, to remove unbound gold complexes and phosphate, and negatively stained with 1% aqueous uranyl acetate (Pella; Redding, CA) containing 0.2% tylose for 1 min (Hayat 1981). After labeling, the grids were blotted on Whatman number 1 filter paper and allowed to air-dry overnight in a grid box. Gold particles were viewed on a Zeiss 10A transmission electron microscope (TEM) operating at 100 kV to determine the absolute number of apparent antigenic sites present in a given area of SR membrane.
Double immunogold labeling was performed in some studies by incubating grids with a second primary antibody (i.e., anti-PK) diluted 1:250 in PBS after the first molecule (usually Ca2+-ATPase) was labeled with primary and secondary antibodies. After 30-min incubation, grids were rinsed with PBS and then incubated for 30 min with a second secondary antibody (e.g., donkey anti-goat IgG) conjugated to 12-nm colloidal gold diluted 1:40 in PBS. Grids were then washed, stained, and dried as described above. In some studies the order of labeling of the Ca2+-ATPase and PK molecules was reversed. PK was labeled first with 6-nm gold particles and Ca2+-ATPase was labeled second with 12-nm particles.
Thin Section On-grid Labeling
Cardiac SR vesicles were pelleted by an Eppendorf centrifuge at 12,000 rpm for 5 min and suspended in 1.5 ml of PBS containing 2% paraformaldehyde and 0.05% glutaraldehyde. Samples were microwave-pulsed with 10 sec pulse/ 20 sec rest/10 sec pulse at 100% power, then briefly rinsed in PBS followed by 0.1 M sodium cacodylate, both containing 50 mM ammonium chloride, and stained in 1% uranyl acetate for 15 min. Pellets were quickly dehydrated through a graded series of ethanol (50, 70, and 90%), then infiltrated with Eponate (Pella) and cured overnight at 50C. Ultrathin sections (70–90 nm) of cardiac SR vesicles were cut on a low-angle Diatome knife and subsequently placed on the 200-mesh nickel grids. Immunogold labeling was performed as described above. After the labeling, all sections on grids were contrasted with lead citrate and examined in a Zeiss 10A TEM operating at 80 kV.
Quantitative Electron Microscopic Analyses
The apparent immunogold labeling density was determined by counting all of the particles on the surface of SR vesicles in electron micrographs printed at a final magnification of X 154,000–308,000. The bottom of each vesicle was considered to adhere to the grid and therefore to be inaccessible for immunolabeling. Vesicles were assumed to have become flattened during processing (see folding of vesicle surface apparent in Figures 1, 2, and 4–7), leaving a circular or elliptical surface exposed for labeling. The apparent labeling density (D) was calculated as
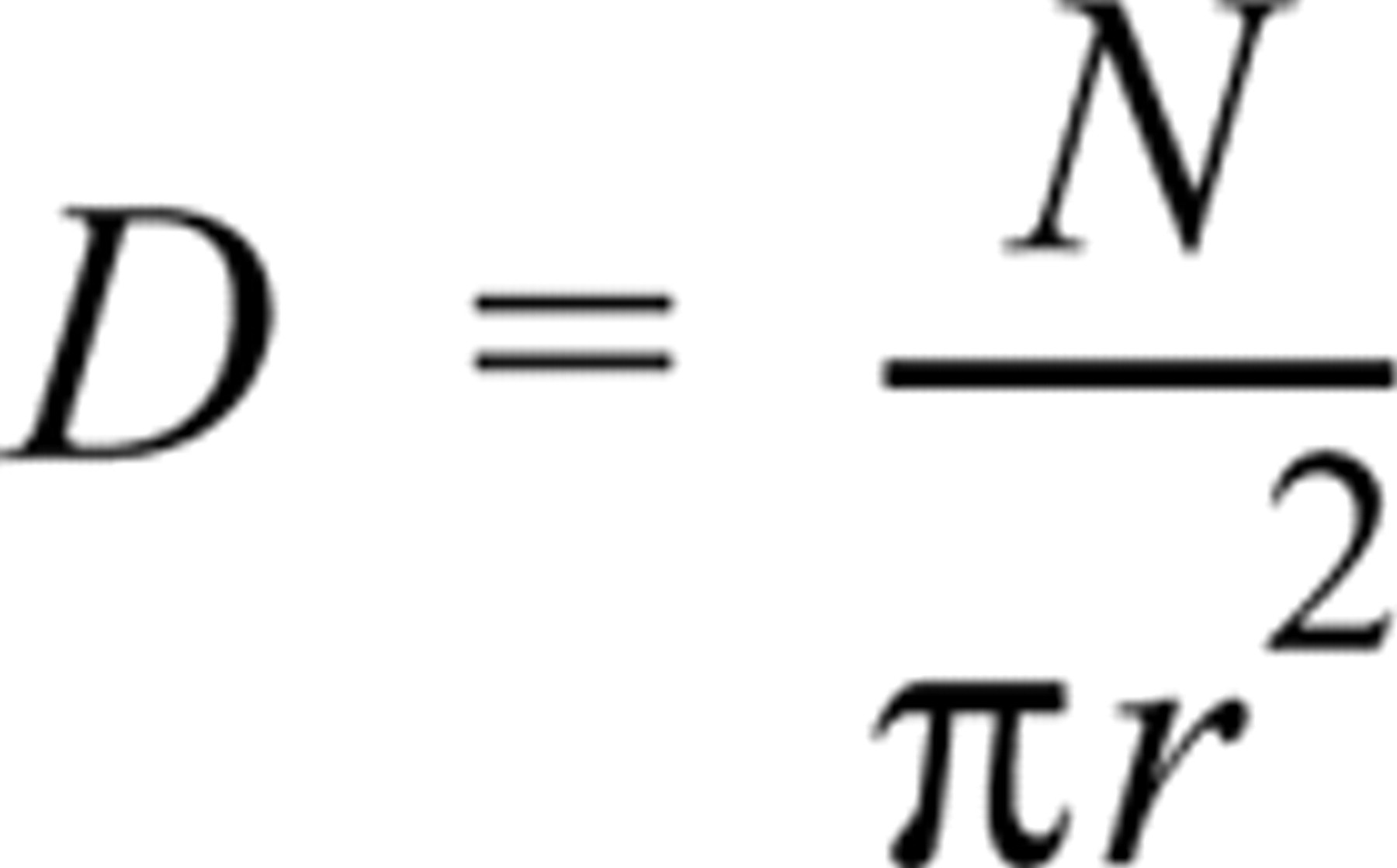
or
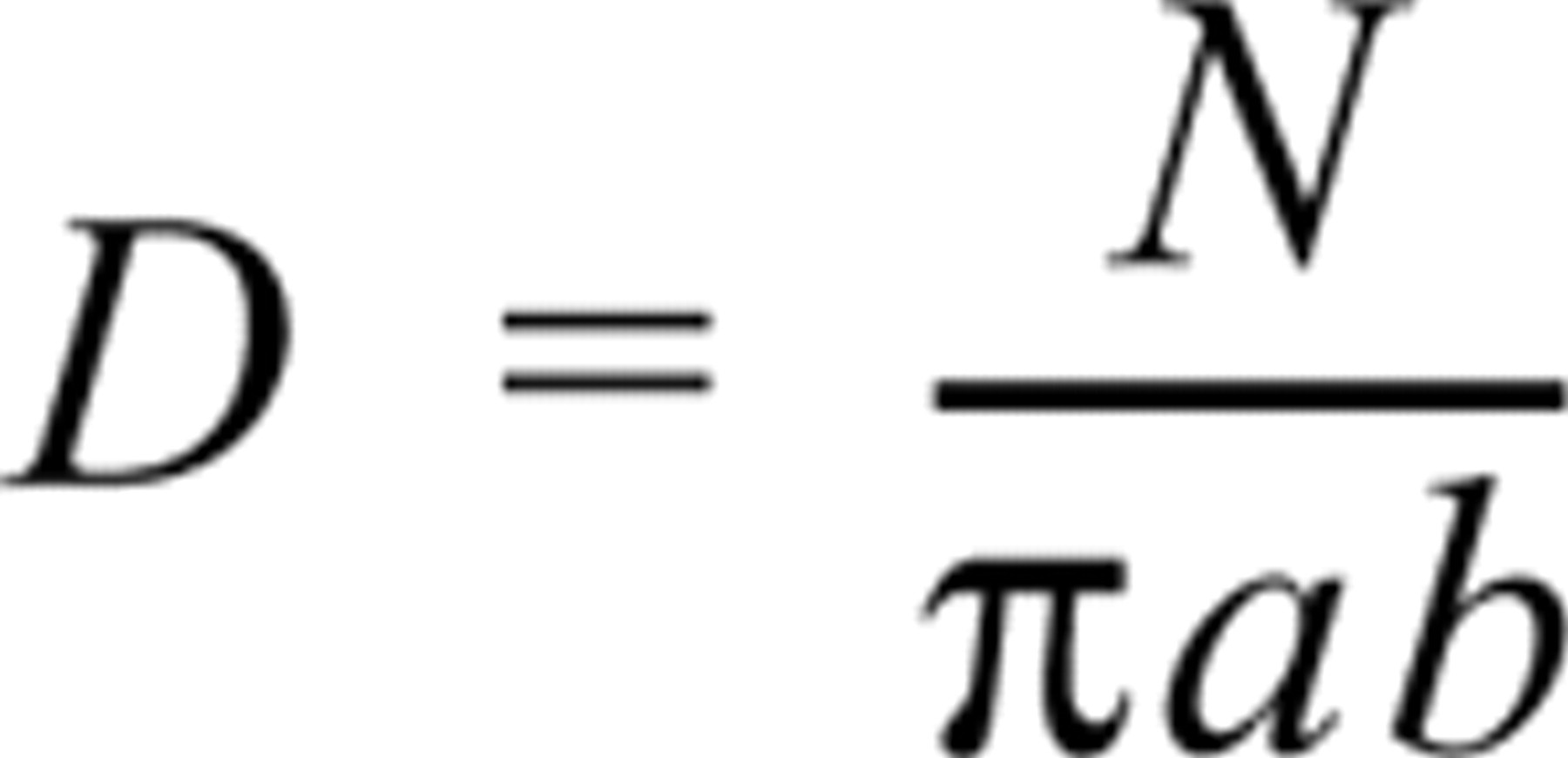
where N is the total number of particles present on a vesicle and r is the radius of a circular vesicle, and a and b are the major and minor semi-axes of an elliptical vesicle. Labeling density was expressed as the number of gold particles per μm2 of SR surface area. The data were corrected for background labeling (less than 0.01 particles/μm2; data not shown). Each data point represents the mean of five to ten independent electron micrographs. Values cited are the mean ± SD.
Results
Ultrastructural Localization of PK
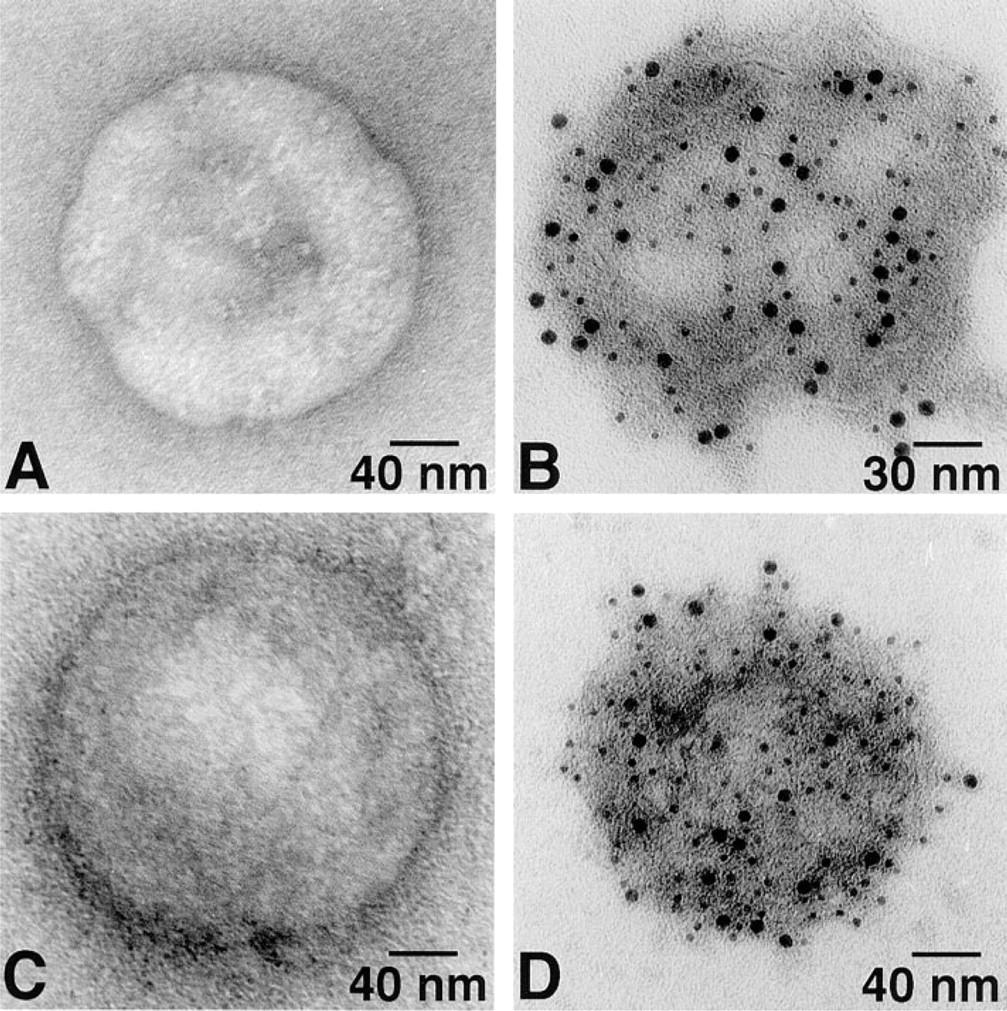
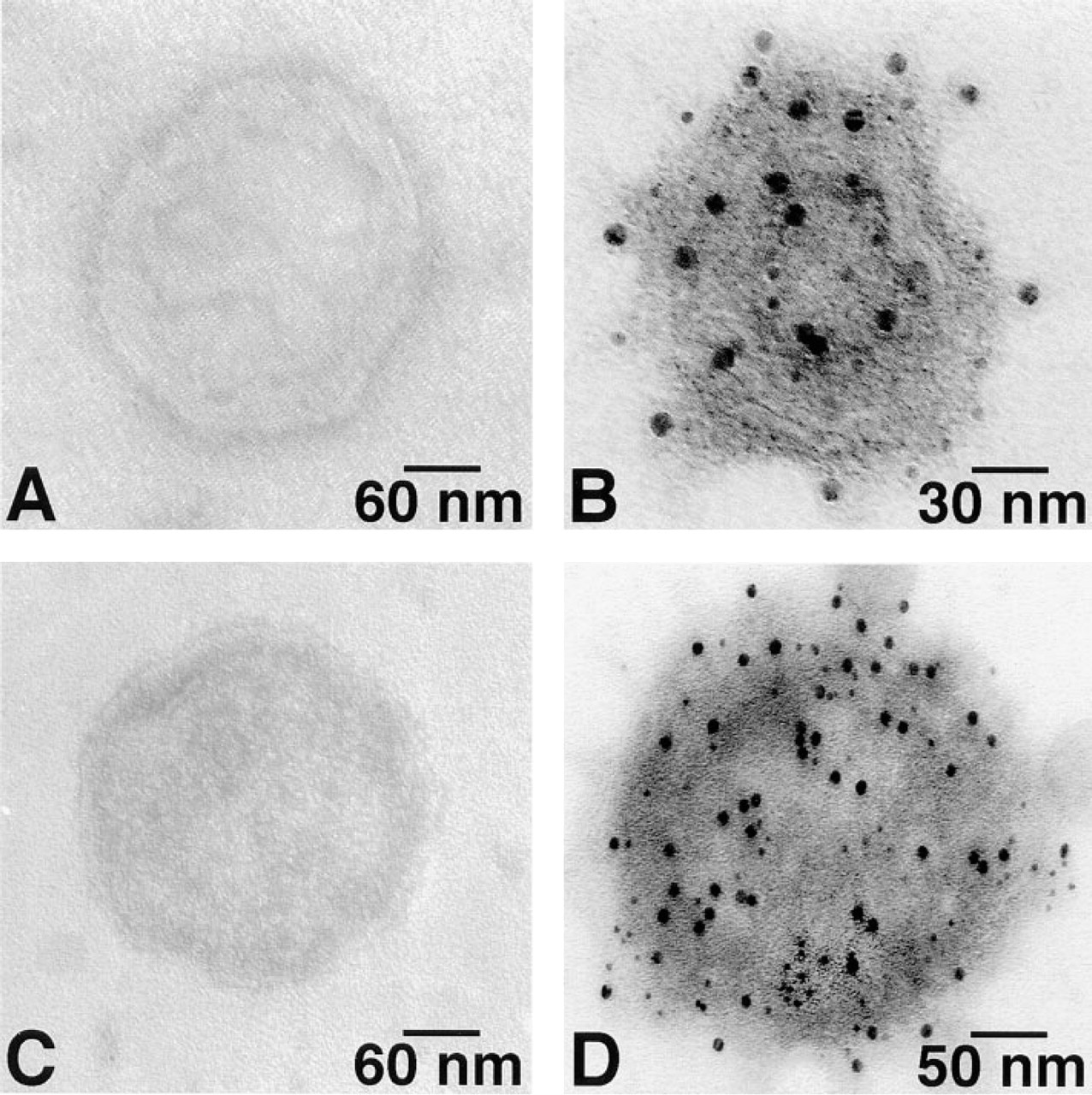
Ca2+-ATPase and PK were broadly dispersed on the surface of cardiac SR vesicles with both 6- and 12-nm gold probes (Figure 1). The same random distributions of Ca2+-ATPase and PK were also found on skeletal muscle SR vesicles (Figure 2). No labeling occurred in the absence of primary antibody (Figures 1A, 1D, 2A, and 2D). As summarized in Figure 3, after labeling of PK with 6-nm gold particles the number of particles/μm2 was 2522 ± 445 and 4171 ± 1379 for cardiac and skeletal muscle SR, respectively. For 12-nm gold particles, the values were 1469 ± 830 and 2619 ± 1000, respectively. After labeling of Ca2+-ATPase with 6-nm particles, densities of 2550 ± 639 and 3877 ± 408 particles/μm2 were observed for cardiac and skeletal muscle SR, respectively. For 12-nm particles, the values were 1420 ± 563 and 2636 ± 750 particles/μm2. The labeling densities of SR Ca2+-ATPase and PK were approximately equal in both cardiac and skeletal muscle SR, but skeletal muscle SR had 60–80% more labeling than cardiac SR for both molecules. Six-nm gold particles provided greater labeling density than 12-nm particles for both types of SR vesicles and for both PK and Ca2+-ATPase, suggesting that the larger probe may have had a lower labeling efficiency owing to a steric hindrance effect.
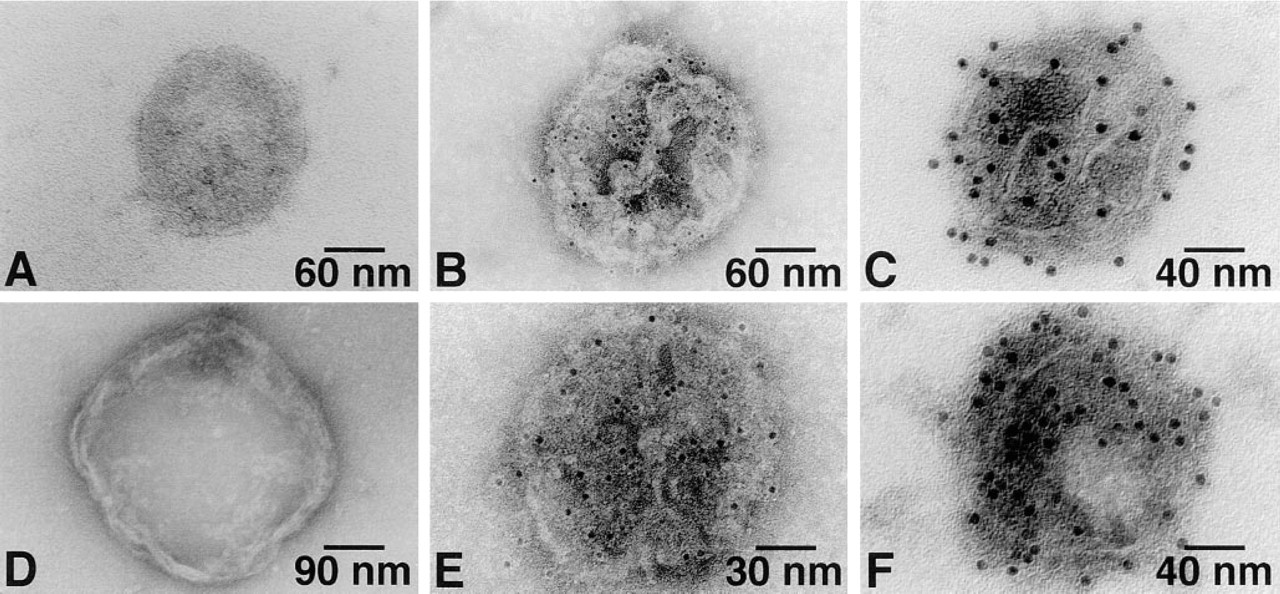
Single immunogold labeling of the cardiac SR vesicle Ca2+-ATPase (
Ultrastructural Co-localization of PK with Ca2+-ATPase
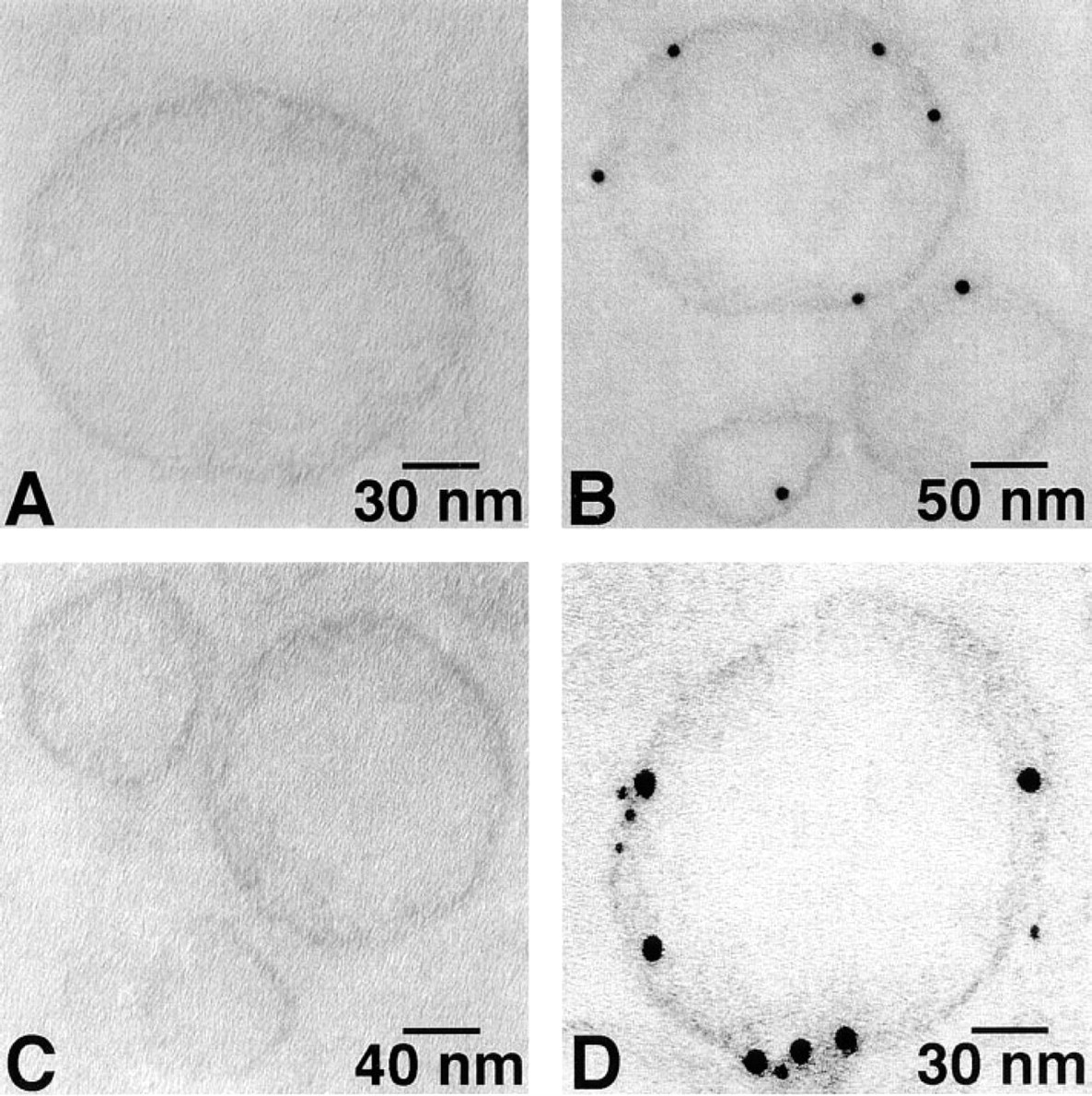
SR vesicles were immunolabeled with antibody against Ca2+-ATPase using the 6-nm gold probe and then with antibody against PK using the 12-nm gold probe. These double immunogold labeling studies clearly showed that PK is co-localized with Ca2+-ATPase on the cytoplasmic surface of both types of SR vesicles (Figures 4B and 4D).
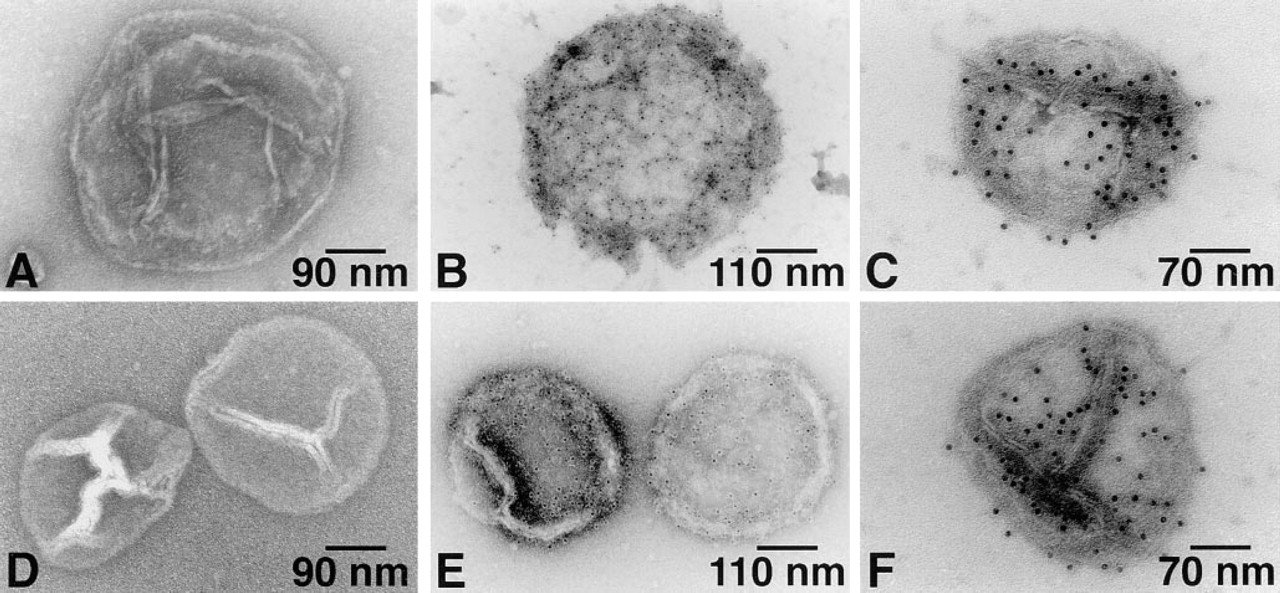
Single immunogold labeling of skeletal muscle SR vesicle Ca2+-ATPase (
Distribution of Pyruvate Kinase on Ultrathin Sections
Immunolabeling of PK performed on ultrathin sections (70–90-nm thickness) of cardiac SR vesicles demonstrated that the colloidal gold particles were apparently attached to the SR membrane (Figure 5B). In double labeling studies, particles marking PK and Ca2+-ATPase molecules appeared to be located immediately adjacent to one another (Figure 5D).
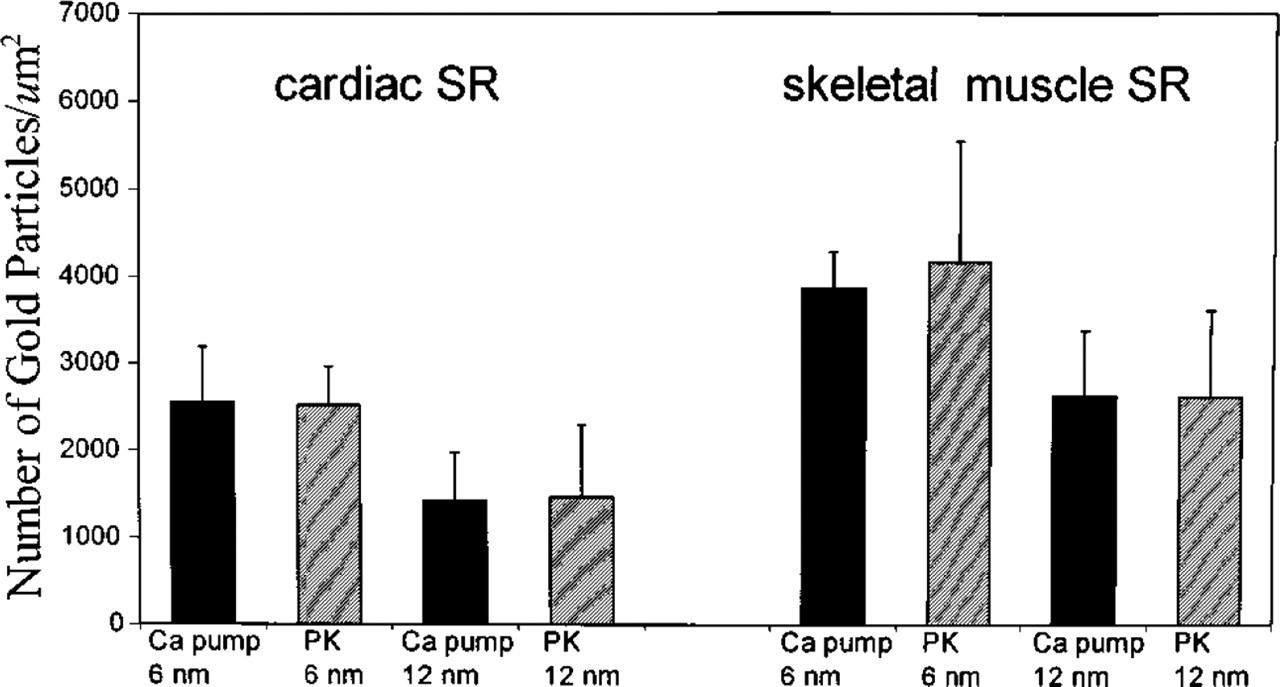
The apparent labeling densities of gold particles (num-ber/μm2) of SR Ca2+-ATPase and PK were approximately of equal molar ratio. The data represent the mean ± SD of five to ten independent electron micrographs.
Presence of Aldolase and GAPDH on SR Vesicles
Using the same double immunogold labeling methods, we also identified aldolase and GAPDH on SR vesicles. Both molecules appeared to be distributed randomly on cardiac (Figure 6) and skeletal muscle (Figure 7) SR vesicles. The specificity of the labeling for these glycolytic enzymes was demonstrated by the lack of binding in the absence of primary antibodies (Figures 6A, 6C, 7A, and 7C).
Discussion
Indirect evidence exists that glycolytic enzymes are associated with SR and are functionally coupled to the SR calcium pump (Xu et al. 1995). We have previously shown that addition of glycolytic substrates and co-factors to isolated SR vesicles from cardiac or skeletal muscle produces sufficient ATP to support Ca2+-ATPase activity and 45Ca transport. Furthermore, the ATP produced by glycolysis in isolated SRs is relatively protected from a soluble ATP trap and is more effective at supporting Ca2+-ATPase activity and 45Ca transport than exogenously supplied ATP.
This study was designed to determine whether anatomic localization of ATP-producing glycolytic enzymes on SR membranes could underlie the apparent functional coupling between glycolytic ATP and SR calcium transport. Using electron microscopy with single and double immunogold labeling of Ca2+-ATPase and several glycolytic enzymes, we found that PK, aldolase, and GAPDH, as well as Ca2+-ATPase, are all broadly dispersed on SR membranes from both cardiac and skeletal muscle. This is the first study to directly demonstrate that several important glycolytic enzymes are bound to SR in close proximity to the Ca2+-ATPase (calcium pump). Our findings provide an anatomic basis for the observation that ATP generated by SR-associated glycolytic enzymes is functionally coupled to SR calcium transport.
Double immunogold labeling of rabbit cardiac (
It is known that most isolated SR vesicles show a right side out configuration (Clarke et al. 1990). The monoclonal anti-SERCA1 and anti-SERCA2 antibodies are specifically directed against the cytoplasmic portions of the skeletal and cardiac muscle SR Ca2+-ATPase. The EM data presented here therefore indicate that PK, aldolase, and GAPDH are likely to be localized on the cytoplasmic surface, as is the epitope of SR Ca2+-ATPase.
Several technical issues should be addressed. The adequacy of molecular labeling depends on the specificity and affinity of the antibody used in the first step of the immunolabeling technique. All of the antibodies purchased for this study were well characterized (Affinity BioReagents and Jackson ImmunoResearch Laboratories) and were of the highest immunological specificity. To block potential nonspecific binding (mainly due to free aldehyde groups), every grid was immersed in a solution of 5% (w/v) BSA in PBS for 10 min before incubation with the primary antibody. In addition, two important controls, omission and substitution of the primary antibody, were performed throughout the study. The colloidal gold-complexed secondary antibodies used in this study recognized only the specific primary antibody, because no gold particles were seen either in the absence of primary antibody (Figures 1–5), or with an irrelevant antibody against the β-subunit of the Na+,K+-ATPase (data not shown). Furthermore, the double labeling controls also indicated that neither secondary gold-labeled antibody bound to the other primary antibody (data not shown).
A common limitation of EM immunogold labeling is steric hindrance, related to gold probe size, and multiple antibody molecules binding noncovalently to each gold particle (two to four antibodies/6-nm gold and six to ten antibodies/12-nm gold; communication with Jackson ImmunoResearch Laboratories). For detection of SR surface-located proteins, steric hindrance may (a) interfere with the reaction between primary and secondary antibody, (b) prevent access for identifying the second specific antigenic site after the first site is labeled, or (c) affect the labeling pattern, resulting in differences in labeling density. Our results showed that the labeling density of PK and Ca2+-ATPase with 6-nm gold probes was greater than with 12-nm probes for both cardiac and skeletal muscle SR (Figure 3). Similar differences in labeling density among 1-nm, 5-nm, and 10-nm particles have also been reported by other laboratories (Yokota 1988; Dulhunty et al. 1993). In addition to steric hindrance, other technical problems may arise. The antigenic conformation of the Ca2+-ATPase or PK could be damaged during isolation of SR vesicles, resulting in areas of nonlabeling on the SR surface and nonhomogeneous distribution along the outer SR membrane. For ultrathin section labeling, the target molecules may be denatured by the glutaraldehyde prefixation, preventing recognition by the corresponding antibody. For all of these reasons, it is unlikely that all of the target molecules on the SR surfaces will be labeled.
Electron micrographs of thin-sectioned cardiac SR vesicles, to which the colloidal gold-conjugated secondary antibody was added after the incubation with primary antibody. (
In skeletal muscle SR, a density of 4000–6000/μm2 has been reported for Ca2+-ATPase using freeze-fracture techniques (Beringer 1976; Heilmann et al. 1981) and 4310/μm2 in pig gracilis fibers and the terminal cisternae of rat extensor digitorum longus muscle using immunogold labeling (Dulhunty et al. 1993). These results are comparable to our finding of 3877 ± 408/μm2 Ca2+-ATPase molecules for rabbit skeletal muscle SR using immunogold labeling. However, using rotary shadowing techniques, a value of 31,000–34,000 Ca2+-ATPase molecules/μm2 was reported for nonjunctional SR (Franzini-Armstrong and Ferguson 1985). Some of the membrane particles observed in this study could have represented proteins other than Ca2+-ATPase molecules. It has been proposed (Taylor et al. 1988) that Ca2+-ATPase forms dimers on isolated SR vesicles. Although no doublet gold particles were seen in our immunolabeling studies, steric hindrance would probably have prevented both molecules of a dimer from being individually labeled. Under our experimental conditions, the density of Ca2+-ATPase labeling is significantly greater on SR vesicles from skeletal muscle than from cardiac muscle (3877/μm2 vs 2550/μm2), consistent with the known dominant calcium pump concentration and higher rates of SR Ca2+-ATPase active transport in skeletal muscle.
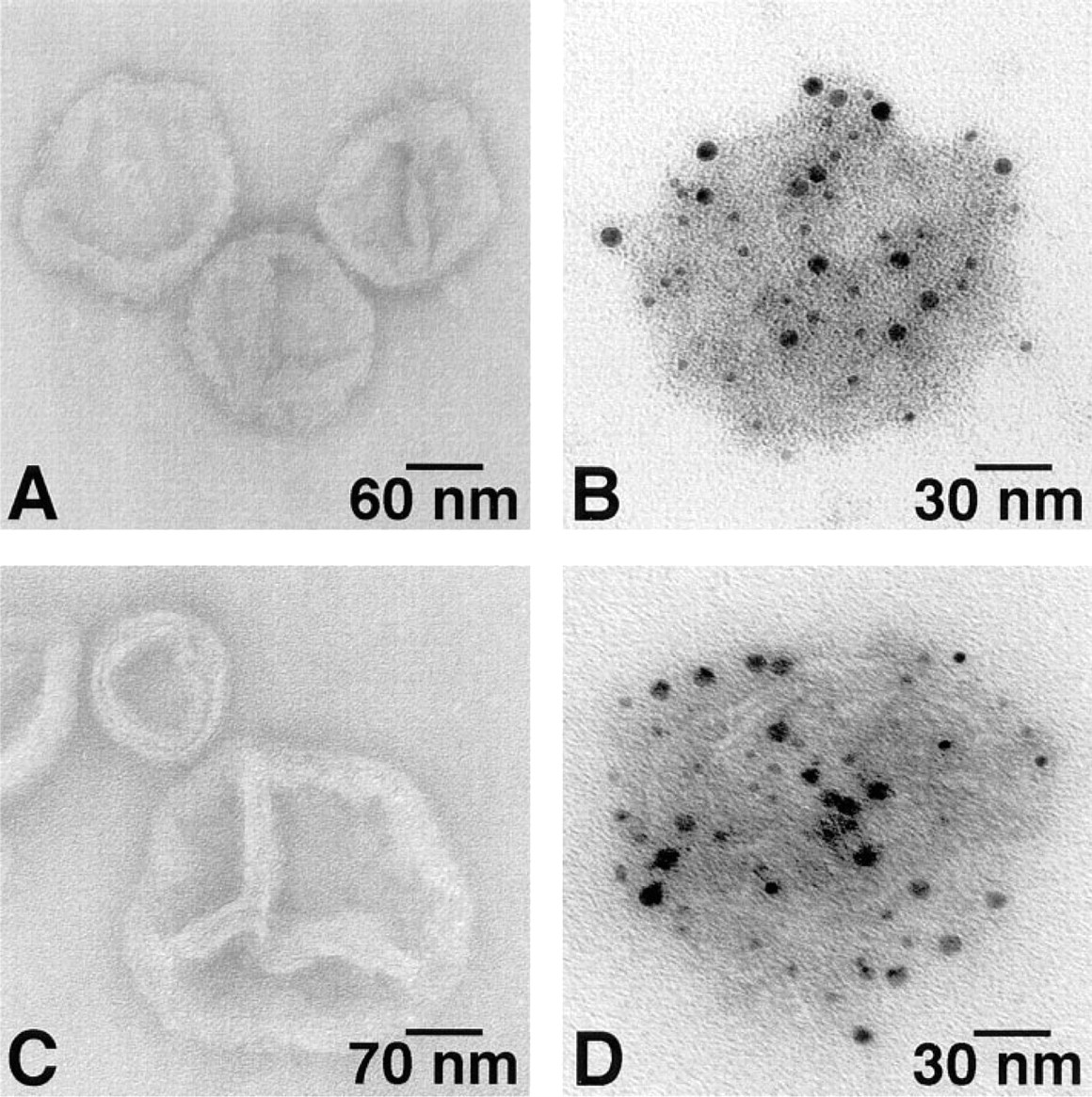
Double immunogold labeling of aldolase (AL) and GAPDH on cardiac SR vesicles. (
It is well known that SR Ca2+-ATPase is an integral membrane protein (Hasselbach and Elfvin 1967; MacLennan et al. 1971; Inesi and Scales 1974; Stewart and MacLennan 1974; Hidalgo and Ikemoto 1977). However, the nature of the way in which glycolytic enzymes are associated with SR membranes is unknown. Our observation (Xu et al. 1995) that glycolytic enzyme activity was lost when isolation buffer containing 600 mM KCl was used is consistent with the report of Pierce and Philipson (1985), who found that GAPDH and phosphoglycerate kinase (PGK) activities were lost from a rabbit cardiac particulate fraction containing SL and SR vesicles in the presence of a high concentration of NaCl. However, the activities were fully recoverable in the supernatant, indicating that the enzymes had been solubilized from the membranes. These results suggest that the binding of at least GAPDH and PGK to SR and SL membranes may be electrostatic in nature and potentially reversible. Electrostatic interactions have also been invoked to explain the binding of hexokinase to mitochondrial membranes in brain and tumor cells (Bustamante and Pedersen 1980; Kabir and Wilson 1993) and of hemoglobin to band 3 in erythrocyte membranes (Low 1989). Ultrathin sections revealed that the gold particles specifically labeled both Ca2+-ATPase and PK along the SR membrane, implying that PK is bound to the SR vesicle membrane. Whether such association is through electrostatic interactions, protein-protein interactions (Ca2+-ATPase to PK, PK to cytoskeletal proteins), or protein-lipid interactions remains to be answered.
In summary, the present studies provide direct ultrastructural evidence that the glycolytic proteins PK, aldolase, and GAPDH are bound to the cytoplasmic side of cardiac and skeletal muscle SR. These results provide an anatomic basis for our previous indirect observations that the ATP generated by SR-associated glycolytic enzymes is coupled to SR calcium active transport. We speculate that the glycolytic enzyme chain may constitute an integral part of the SR Ca2+ transporting system.
Double immunogold labeling of aldolase (AL) and GAPDH on skeletal muscle SR vesicles. (
Footnotes
Acknowledgements
Supported by US Public Health Service grants HL33360, HL52315 and HL52175 from the National Heart, Lung, and Blood Institute, Bethesda, MD.
We are grateful to Ms J. Li and Mr M. Delannoy for carrying out the immunogold labelings.