
Abstract
Recently, we developed the terminal deoxynucleotidyl transferase (TdT)–immunogold technique for in situ detection of DNA molecules. In this study the potential value and the limitations of the method were evaluated using the giant polytene chromosomes from Chironomus tentans salivary glands. Emphasis was put on the Balbiani rings (BRs), specialized chromosomal sites with exceptionally intense synthesis of large mRNA molecules. Immunolabeling was recorded not only over the bands and interbands of the polytene chromosomes but also over the BR structures. In the BRs, gold particles were present over segments of active transcription units, each with a central chromatin axis and a number of growing RNP products attached to the axis. One third of the transversely sectioned transcription units showed labeling in the central parts, i.e., where the unfolded chromatin axis is located, whereas the growing RNP fibers remained unlabeled. The absence of labeling of the RNP fibers is not likely to be due to lack of accessibility, because anti-RNA antibodies readily decorated the RNP fibers. The nuclear sap and cytoplasm displayed no significant label. These results clearly indicate that the TdT–immunogold technique is specific for DNA and detects not only DNA in compacted chromatin but also fully extended DNA. Its ability to efficiently label a single DNA molecule demonstrates the method's very high sensitivity.
A
To further elucidate the question of specificity and sensitivity, we chose to investigate the Balbiani rings (BRs) of the giant polytene chromosomes of Chironomus tentans salivary glands. The BRs are sites at which large mRNA molecules are actively produced (Lamb and Daneholt 1979; Daneholt 1975, 1982). They also constitute the only model in which EM techniques, thin sectioning, and Miller-type spreads have all been used to investigate the chromatin, in detail, and particularly the transcription complexes of a given gene. A typical stained section through the BR region usually reveals dozens of short segments of transcription unit loops, each with a thin chromatin axis studded with many electron-dense nascent RNP particles. In the promoter–proximal part of the active gene, the transcription products appear as RNP fibers of growing length along the gene. Further downstream the peripheral ends of the growing RNP fibers are packed into a globular structure; stalked RNP granules are formed. The completed product is an RNP granule, 50 nm in diameter, the BR granule. With our system we can evaluate the sensitivity of the technique by studying chromatin on several well-defined organizational levels, from highly compacted DNA (DNA packing ratio about 400) to superactive, pilocarpin-treated glands (DNA packing ratio close to 1).
In this study we used the in situ terminal deoxynu-cleotidyl transferase (TdT)–immunogold method to investigate the precise location of DNA in this model. This recent technique combines a molecular biology procedure with an immunocytological method (Thiry 1992c). It presents two essential advantages. First, it is compatible with various fixation and embedding conditions and therefore offers the possibility of studying, with high resolution, the precise location of DNA in very well-preserved structures. Second, it allows the detection of DNA-containing structures in a wide variety of organisms, from prokaryotes to eukaryotes, even when the structures are present in very low amounts. Examples of high-resolution detection include the DNA present in mitochondria, chloroplasts, mycoplasmas, and viruses (Thiry 1992b; Thiry and Puvion–Dutilleul 1995). Our results clearly demonstrate the ability of the TdT–immunogold method to detect DNA not only in highly compact chromatin but also in the extended axis of a single active BR gene.
Materials and Methods
EM Preparations
Fourth instar larvae were decapitated and the salivary glands were isolated. Small pieces of various Chironomus tentans salivary glands were fixed for 2 hr at 4C in 2% glutaraldehyde dissolved in 0.1 M sodium cacodylate-HCl buffer (pH 7.2). The pieces were then rinsed four times with the same buffer for 15 min. Next, they were dehydrated in a graded ethanol series and soaked for 15 min at room temperature (RT) in ethanol:Agar 100 resin (3:1), for another 15 min in ethanol:Agar 100 resin (2:1), overnight in ethanol:Agar 100 resin (1:1), then finally embedded in Agar 100 resin (Agar Scientific; Essex, UK) and polymerized first at 45C, then at 60C. The glands were sectioned with a diamond knife in a Reichert Ultracut ultramicrotome. Optimal BRs were located by light microscopy on thick sections stained with toluidine blue. Subsequently, ultrathin sections were collected in platinum rings (4-mm diameter) formed by a platinum wire (0.1-mm in diameter; SA Johnson Matthey, Bruxelles, Belgium) and stored in distilled water until used.
TdT–Immunogold method
Ultrathin sections were first floated for 60 min at RT on a saturated solution of sodium metaperiodate, then incubated for 10 min at 37C at the surface of the following medium: 20 μM 5 bromo-2-deoxyuridine (BUdR) triphosphate (Sigma; St. Louis, MO), 100 mM sodium cacodylate (pH 7), 2 mM MnCl2, 10 mM β-mercaptoethanol, 50 μg/ml BSA, 125 U/ml calf thymus TdT (Boehringer Mannheim; Mannheim, Germany). (Thiry 1992c). Sections were again incubated for 10 min at 37C on the same medium supplemented with 4 μM each of dCTP, dGTP, and dATP (Gibco BRL; Merelbeke, Belgium). After two rinses in double-distilled water, the various sections were incubated for 30 min in PBS (0.14 M NaCl, 6 mM Na2HPO4, 4 mM KH2PO4, pH 7.2) containing normal goat serum (NGS) diluted 1:30 and 1% BSA, then rinsed with PBS containing 1% BSA. The next step of the treatment was a 4-hr incubation at RT with monoclonal anti-BUdR antibody (Becton Dickinson; Mountain View, CA) diluted 1:50 in PBS containing 0.2% BSA and NGS diluted 1:50. After washing with PBS containing 1% BSA, the sections were incubated at RT for 1 hr with gold-coupled goat anti-mouse IgG (particle diameter 5–10 nm; Janssen Life Sciences, Beerse, Belgium), diluted 1:40 with PBS (pH 8.2) containing 0.2% BSA. After washing with PBS containing 1% BSA, the sections were rinsed in deionized water.
Several kinds of control experiments were carried out. First, TdT or labeled nucleotides were omitted from the TdT medium. In a second control, BUdR triphosphate was replaced by BUdR monophosphate. Third, the grids were incubated with antibody-free particles. In the fourth control, the primary antibody was omitted. Finally, some cell sections were preincubated at 37C for 120 min with 1 mg/ml DNase I (Sigma; Type DN-Ep) in PBS (0.14 M NaCl, 6 mM Na2 HPO4, 4 mM KM2 PO4, pH 6.8) containing 7 mM MgCl2. Other sections were preincubated at 56C for 120 min with 1 or 10 mg/ml RNase A (Boehringer Mannheim) in 10 mM Tris-HCl (pH 7.4) containing 15 mM NaCl.
Immunocytological Technique for RNA
The procedure, involving two mouse monoclonal anti-RNA antibodies (D444, BWR5), has been described (Thiry 1992a). For labeling, ultrathin sections of Agar-embedded cells were incubated for 25 min in PBSB (pH 7.2) containing NGS and normal rabbit serum (NRS), each diluted 1:30, then for 3 hr at RT in RNA-specific antibodies diluted 1:10 in PBSB containing NGS and NRS, each diluted 1:50. After five rinses in PBSB, the sections were incubated for 30 min with goat anti-mouse IgG3 (heavy-chain specific; Sigma) diluted 1:100 in PBSB containing NGS and NRS, each diluted 1:50. After four rinses in PBSB (pH 7.2) plus one in PBSB (pH 8.2), sections were transferred to an incubation medium containing gold-coupled rabbit anti-goat IgG (particle diameter 5 nm; Janssen Life Sciences) diluted 1:50 in PBSB (pH 8.2). Incubation was for 60 min at RT. Samples were then rinsed with PBSB, followed by distilled water.
Control experiments were carried out as previously described (Thiry 1992a). When the primary or secondary antibody, or both, were omitted, the ultrathin sections were devoid of label. When the grids were incubated with antibody-free particles, no labeling occurred. No label was detected when sections were preincubated with RNase A.
Finally, the ultrathin sections were mounted on nickel grids and stained with uranyl acetate and lead citrate before examination in a JEOL CX 100 electron microscope at 60 kV.
Quantitative Evaluations
To evaluate the labeling density, we first determined the area (Sa) of each compartment morphometrically by the point-counting method (Weibel 1969). We then counted the number of gold particles (Ni) over each compartment and calculated the labeling density (Ns = Ni/Sa). To evaluate DNA labeling densities on salivary gland cells fixed under isotonic conditions, we analyzed 25 random micrographs and counted 13,023 gold particles. One should bear in mind that these numerical data do not reflect the exact numbers of DNA molecules in the compartments studied but merely the relative densities of the different compartments.
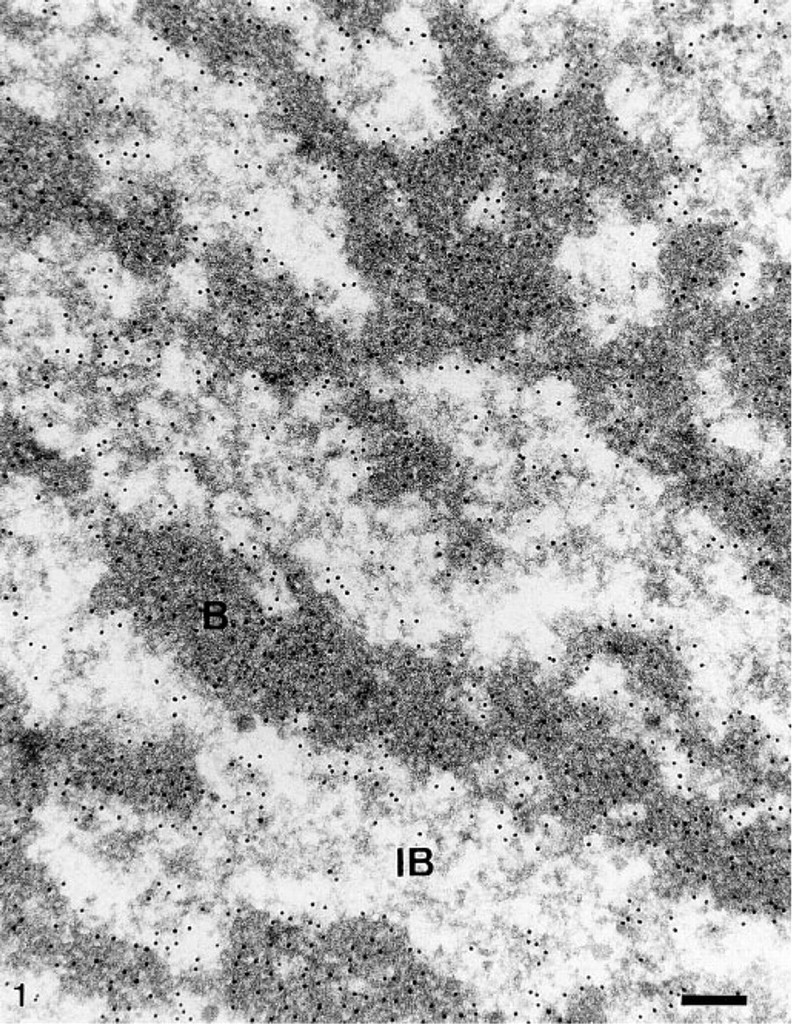
Distribution of DNA in polytene chromosomes revealed by the in situ TdT–immunogold method. Parts of electron-dense bands (
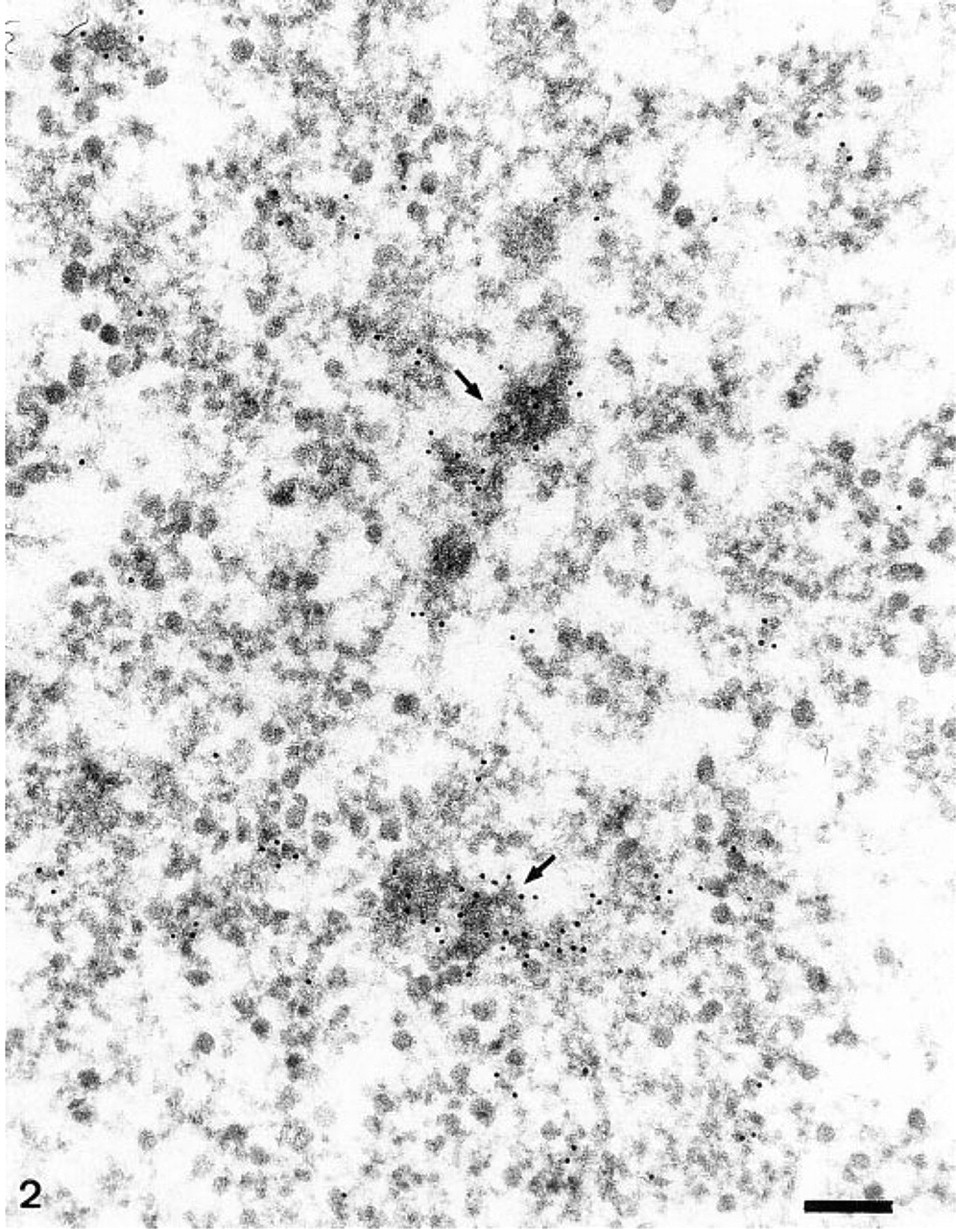
Distribution of DNA in a Balbiani ring studied with the in situ TdT–immunogold method. Gold particles are scattered over the various segments of active BR genes. Intense label is found over two chromosomal branches of condensed chromatin (arrows). Bar = 0.2 μm.
Results
BR Structures Contain DNA-positive Sites
To pinpoint the location of DNA within the polytene chromosomes of Chironomus tentans salivary glands, and notably within the BRs, we applied the TdT–immunogold procedure to ultrathin sections. In this procedure, free DNA ends generated by sectioning are elongated by the enzyme TdT in the presence of bromodeoxyuridine triphosphates. The thymidine analogue is visualized by immunogold electron microscopy. The micrographs were examined visually and evaluated quantitatively. Under our experimental conditions, intense label was always found over both the bands and the interbands of the giant polytene chromosomes (Figure 1). The bands were more intensely labeled than the interbands. Evident but less intense label was consistently found over the BR structures (Figure 2), whereas the extrachromosomal areas of salivary gland cell interphase nuclei appeared completely gold-free. Cytoplasmic areas (mitochondria excepted) were devoid of label.
Gold particle densities (number of particles per μm2) over the different structural compartments in Chironomus tentans salivary gland cells fixed under isotonic conditions before TdT–immunogold labelinga
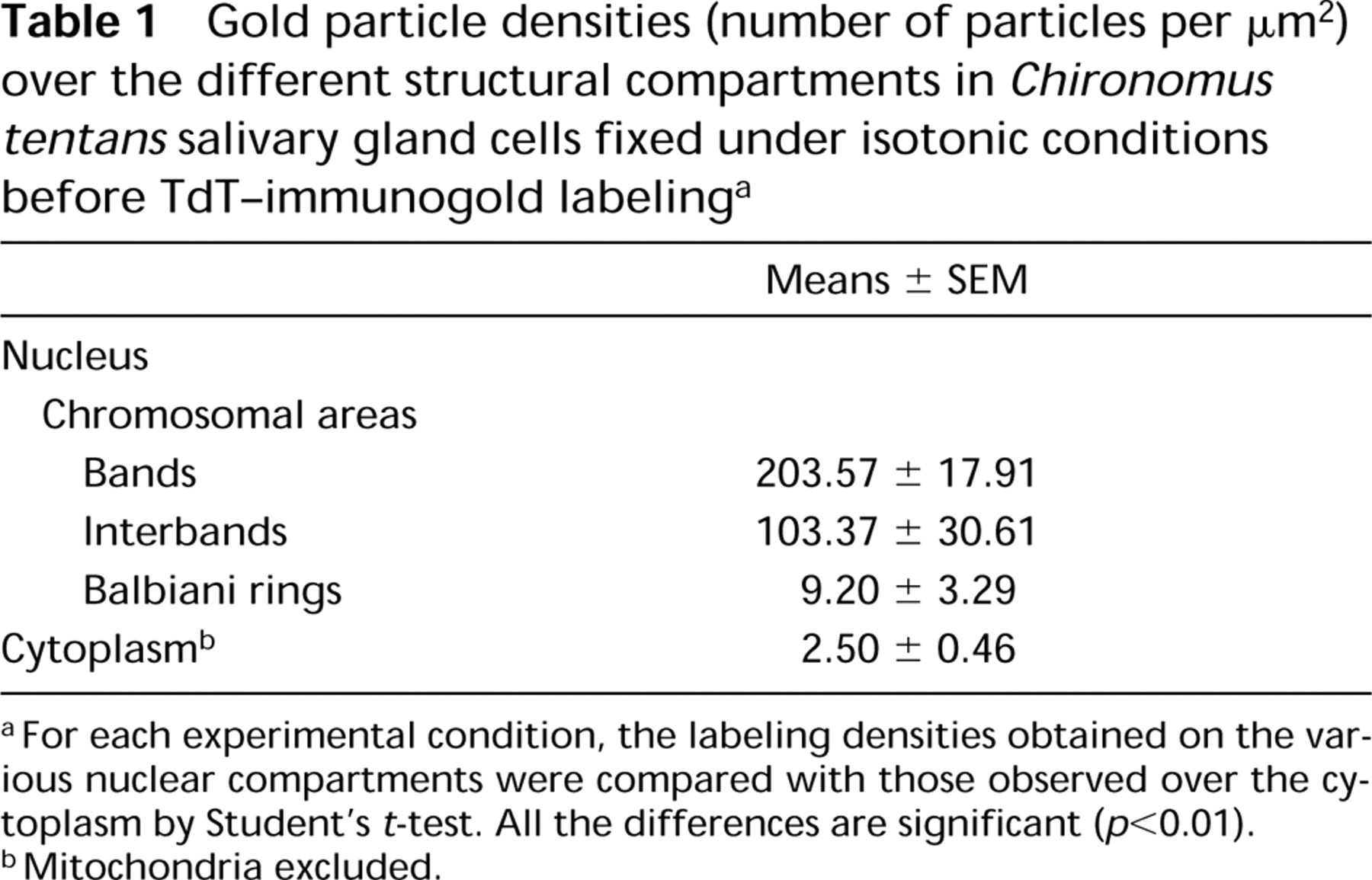
For each experimental condition, the labeling densities obtained on the various nuclear compartments were compared with those observed over the cytoplasm by Student's t-test. All the differences are significant (p<0.01).
Mitochondria excluded.
Table 1 summarizes the numerical data on gold particle distribution over the various compartments. These data confirm our subjective observations and further demonstrate the high labeling specificity. Over the ribosome-rich cytoplasmic areas, the labeling density is insignificant; over the BR structures, it is significant but lower than over areas known for their high DNA content, such as the bands and interbands of chromosomes.
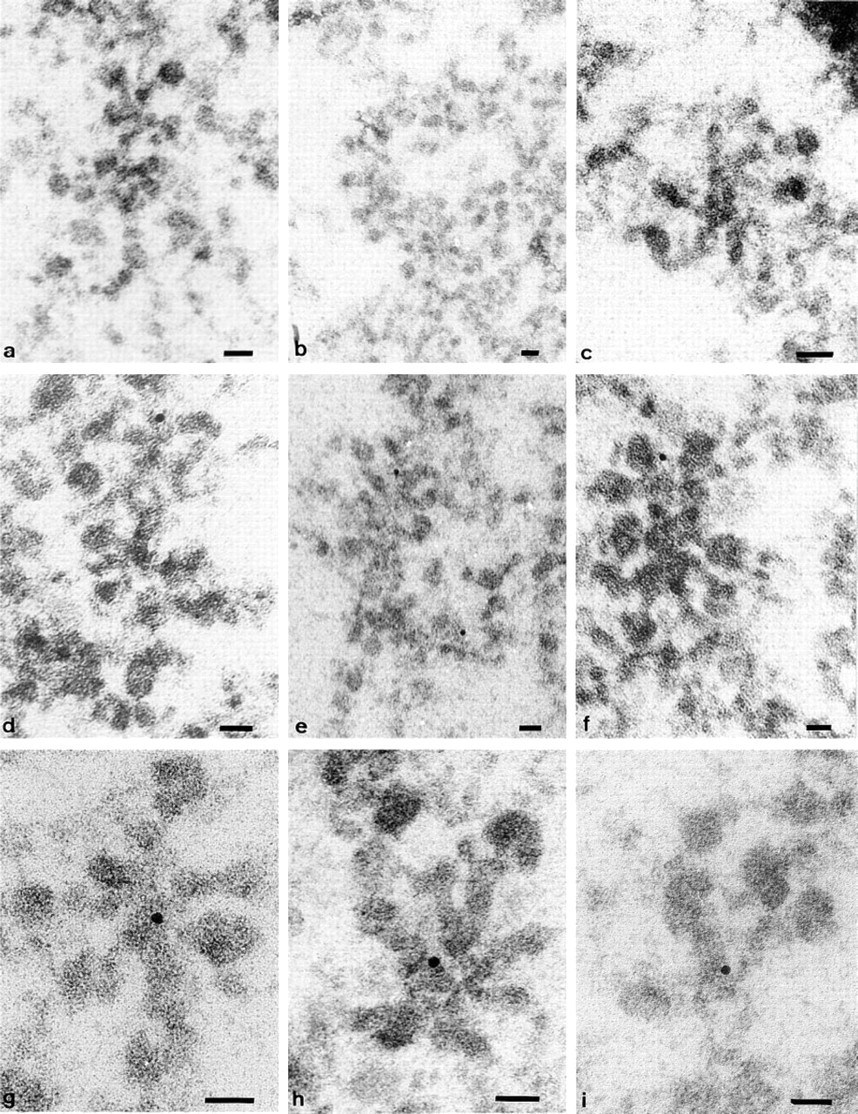
Distribution of DNA within BR substructures as shown by the in situ TdT–immunogold method. Examples of all the types of sections through the active BR genes. Longitudinal sections passing along the active gene exhibit no label (
The specificity of the TdT reaction was tested in several ways. When TdT or labeled nucleotides were omitted from the TdT medium, the ultrathin sections were devoid of label. Likewise, no labeling was observed when BUdR triphosphate was replaced by BUdR monophosphate. When cell sections were incubated with RNase, the labeling persisted. It was almost completely abolished, however, when DNA was specifically digested with DNase I. The immunolabeling specificity was also tested. When the primary antibody was omitted, virtually no labeling occurred. Gold lacking the antibody tag did not bind to the sections.
Detection of DNA in the Axis of a BR Gene
Careful examination of the BR structures shows a “preference” of the gold particles for active transcription unit segments with growing RNP products (Figure 2). Because of intertwining of the segments, it was usually difficult to attribute label to any particular substructure of the loop segments. To achieve this, we had to examine the precise location of label over segments whose axis was separate from and not touching other active genes. A segment's typical labeling pattern was found to depend on the angle at which it was sectioned (Figure 3). It should be recalled that the method detects only DNA ends present at the surface of sections (Thiry 1992c). Longitudinal sections passing along the active gene usually exhibited no label (Figures 3a–3c); the DNA did not reach the surface of the section or is not even within the section. In oblique sections (Figures 3d–3f), positive sites were most often observed over one of the segment extremities, i.e., where DNA is believed to be cut and exposed to the antibody. Finally, transverse sections through the transcription units (Figures 3g–3i), i.e., where DNA is located, consistently displayed gold particles in the central part of the “stars.” This observation was based on the analysis obtained from six different TdT experiments on ultrathin sections (5–17 sections/grid) through 20 different salivary glands sectioned at five different levels. For each of these six experiments, labeled structures were found. The labeling was seen in 32 of 89 structures examined. This corresponds to one third of the transversely sectioned transcription units labeled. Therefore, the method detects the unfolded DNA in the axis of single transcription units.
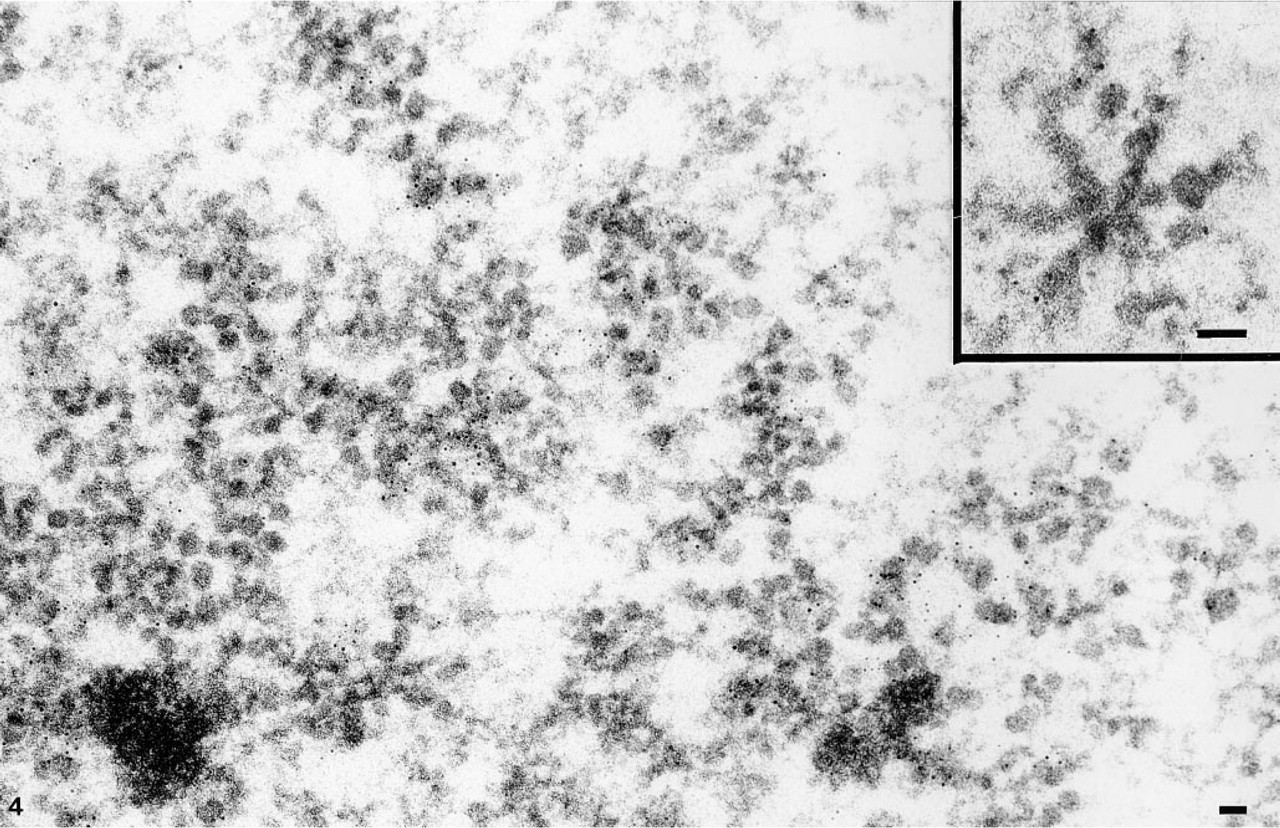
Distribution of RNA within the BRs examined by immunogold labeling with anti-RNA antibodies. Intense label is found over the growing RNP particles in active BR genes. No label is present over chromosomal bands of condensed chromatin. (Inset) A transverse section of an active BR gene. Label is revealed over lateral RNP fibers. Bars = 0.05 μm.
By comparison, the presence of RNA in BR structures was investigated using a postembedding and immunogold labeling with two RNA-specific monoclonal antibodies. Except for the chromosome bands, all the compartments labeled by the TdT procedure were also labeled by this technique. Label was evident over the BR structures (Figure 4). Gold particles were found over almost all active transcription unit segments with growing RNP products, irrespective of the sectioning angle. In particular, in transversely sectioned transcription units, label was revealed over lateral RNP fibers (Figure 4). Label was as abundant over the growing RNP fibers of the proximal segment as over the stalked granules of the distal segment.
Discussion
This study provides considerable information about the potential value of the TdT–immunogold technique in pinpointing the location of DNA in biological materials.
Our results show that the procedures used here can detect DNA in transcriptionally very active genes, such as the 75S RNA genes present in the BRs of C. tentans salivary glands.
The in situ TdT–immunogold technique labels specifically and with high resolution all known DNA-containing structures. In the polytene chromosomes it reveals not only the condensed chromatin of the chromosome bands but also the relatively decondensed chromatin of the interbands. Previous reports have shown the technique to detect both the condensed and dispersed chromatin of interphase nuclei from various tissues and cultured cell lines (Thiry 1992b, c; Thiry et al. 1993). The novelty here is the demonstration that transcriptionally very active chromatin can also be detected by this method. Such active regions of the genome are apparently packaged in an altered nucleo-some structure and are associated with domains at which the chromatin is less condensed or more open than in inactive domains (Reeves 1984). When transcriptional activity is exceptionally high, EM reveals few if any nucleosomes (McKnight and Miller 1976; McKnight et al. 1976; Osheim and Miller 1983), and in nuclease digestion experiments the nucleosomal repeats disappear (Wu et al. 1979; Levy and Noll 1981; Karpov et al. 1984; Widmer et al. 1984). EM studies have shown that, during intense transcription of the BR genes, a large part of the chromatin axis is in an unfolded configuration, i.e., the DNA molecule is more or less fully extended (Andersson et al. 1980; Björkroth et al. 1988). The TdT–immunogold method can therefore identify DNA-containing structures in both inactive and highly active chromatin regions.
Although the restriction of labeling to the surface of the sections is limiting (Thiry 1992c), the TdT immunological procedure is capable of labeling various DNA-containing structures, some containing very low amounts of DNA (e.g., mitochondria, mycoplasmas, virus) (Thiry 1992b,c; Thiry et al. 1993; Thiry and Pu-vion–Dutilleul 1995), which suggests that this technique is very sensitive. We show here that it can reveal DNA present in the unfolded axis of a single active BR gene. Our statistical analysis indicate that in a third of the crossing sections at active BR genes the axis is detected on ultrathin sections. The fact that it is possible to efficiently label a single DNA molecule demonstrates the method's very high sensitivity.
Footnotes
Acknowledgements
Supported by the Fonds de la Recherche Scientifique Médicale (grant no. 3.4516.96). M. Thiry is a research associate of the National Fund for Scientific Research (Belgium).
We are grateful to Prof G. Goessens (University of Liège, Belgium) for encouraging discussions and for critical reading of the manuscript. We also acknowledge the skillful technical and secretarial assistance provided by Ms F. Skivée, Mr D. Bourguignon, and Ms S. Bodson.