
Abstract
Histotechnological processing of DNA can cause damage to and loss of DNA and can change its structure. DNA probes have severe tissue-staining limitations. New DNA probes and improved histotechnology are needed to enhance the characterization of fixed tissue-bound DNA. Our team developed a novel DNA staining technique and histotechnological processing procedure that improves tissue-bound DNA retention and the qualification and quantification of intact double-stranded (ds)-B-DNA. We used the ultrasensitive PicoGreen ds-DNA probe for the histochemical characterization of ds-DNA. Fifteen fixatives were examined to determine which were best for preventing DNA denaturation and retaining original DNA content and structures. Our use of a microwave–vacuum oven reduced heating temperatures, shortened heating and processing times, and enhanced fixation. We achieved better qualitative and quantitative results by using superior tissue-acquisition techniques (e.g., reduced prefixation times) and improved histotechnology. We also compared our novel approach with archival tissues, delayed fixation, less sophisticated and conventional histological processing techniques, and by experimenting with preservation of tissue-bound ds-Z-DNA. Results demonstrate that our histotechnological procedure and nucleic acid staining method significantly improve the retention of intact, undamaged ds-DNA which, in turn, allows the investigator to more precisely quantify the content and structures of unaltered and undamaged tissue-bound ds-B-DNA.
Keywords
DNA
Histochemical, immunohistochemical, laser-capture microdissection, in situ hybridization, in situ polymerase chain reaction, and terminal deoxynucleotidyl transferase-mediated (dUTP) nick end labeling assays require tissues to be properly preserved so the location and physical properties of the antigen remain unaltered and accessible to the nucleic acid probes (Kiernan and Mason 1996; Carson 1997; Bowtell and Sambrook 2003). This requires proper use of the appropriate fixative and histological processing procedure to preserve and retain the fixed nucleic acids. Fixatives must be able to alter the soluble contents of the cell into substances that are insoluble and/or immobile so they are not altered and/or lost during subsequent processing.
Use of a microwave–vacuum oven improves processing procedures, which produces superior tissue sections for molecular studies (Kok and Boon 1992; Marani et al. 1996; Giberson and Demaree 2001). However, the investigator must be aware of all potential factors (e.g., pH, fluid concentrations, embedding media, temperature, environmental contaminants, processing times, light–DNA probe reactions, and purity of fixatives, chemicals, and reagents) before designing a histological–molecular biological project. Certain reagents used for tissue fixation may diminish or completely inhibit the binding ability of a nucleic acid probe. No one fixative has proven to be perfect for the demonstration of all tissue-bound antigens. We believe that to properly quantify intact, unaltered, double-stranded (ds)-DNA content and structure, a molecular biological approach that requires optimal preservation of tissue-bound ds-DNA must be used. Routine methods do not properly conserve the location, DNA content, and structures (e.g., right-handed ds-B-DNA, left-handed ds-Z-DNA) of nucleic acids in fixed tissues.
There are many fixatives available to the investigator. We chose to examine 15 types representing different classes of tissue fixatives (e.g., cross-linking, precipitating, mercury, picric acid, dichromate, osmic acid, slightly modified, and commercial). Our histotechnological processing approach, choice of fixative, and PicoGreen staining technique allow for processing of tissue with optimal retention of unaltered intact ds-DNA. These novel approaches will improve DNA ploidy analysis and diagnostic procedures, as well as the ability to localize and quantify fixed ds-DNA content in cell death studies [e.g., necrosis, apoptosis, and terminal differentiation (denucleation)] and other molecular biological processes (Gagna et al. 1997; Al-Gubory 2005).
DNA and RNA are not static, one-dimensional macromolecules; they are dynamic biomolecules that can adopt many different structures, e.g., right-handed B-DNA and A-RNA and left-handed Z-DNA and Z-RNA (Gagna et al. 1997; Herbert and Rich 1999). Therefore, advancements in histotechnology are necessary to preserve alternative structures of nucleic acids such as B-DNA, Z-DNA, cruciform DNA, B-DNA/ Z-DNA junctions, triplex-DNA, quadruplex-DNA, and hairpin nucleic acids.
Many investigators use probes such as ethidium bromide, propidium iodide, or YO-PRO-1 to quantify DNA. However, for example, unbound ethidium bromide dye has a high intrinsic fluorescence that limits the sensitivity of the tissue assay and binds RNA and single-stranded (ss) DNA (Waring 1965; LePecq and Paloetti 1967; Kral et al. 2005; Guillo et al. 2006). PicoGreen has been shown to exhibit better DNA-binding properties than propidium iodide and YO-PRO-1 (Kral et al. 2005; Guillo et al. 2006). Hoechst stains are selective for ds-DNA quantification but only when certain assay conditions are present. Hoechst 33258 requires high salt concentrations to quantify ds-DNA in the presence of RNA. High salt concentrations can cause conformational changes in DNA helical structure, such as B-DNA being converted to Z-DNA (Herbert and Rich 1999). This can cause quantitative problems relative to different ds-DNA structures. Hoechst stains also require low salt solutions for quantifying ds-DNA in the presence of ss-DNA (Labarca and Paigen 1980). The Feulgen reaction, which is used to quantify DNA content and distribution, does not differentiate between ss-DNA and ds-DNA, causing major quantifying problems. Alternatively, the PicoGreen ds-DNA fluorescent stain is an ultrasensitive ds-DNA probe. PicoGreen fluorescence occurs with an excitation maximum at 480 nm and an emission peak at 520 nm (Ahn et al. 1996; Singer et al. 1997; Pomppanen et al. 2000). Our team developed a novel method to directly use PicoGreen to quantify ds-DNA content in tissue sections. Solution studies using PicoGreen have shown that results are not negatively impacted by proteins, nucleotides, ss-DNA, or RNA (Pomppanen et al. 2000). PicoGreen has been used extensively in solution studies (Ahn et al. 1996; Singer et al. 1997; Murakami and McCaman 1999; Pomppanen et al. 2000; Haugland 2002; Guillo et al. 2006), but never in a histochemical context. PicoGreen is an asymmetrical cyanine stain whose properties include high extinction coefficients, negligible intrinsic fluorescence of the unbound dye, and production of large fluorescence enhancements with high quantum yields upon ds-DNA binding (Murakami and McCaman 1999; Pomppanen et al. 2000; Rengarajan et al. 2002; Guillo et al. 2006). A major advantage of this probe is that it remains stable with photobleaching compared with other probes, which allows for longer exposure times (Ahn et al. 1996). PicoGreen dye exhibits > 1000-fold fluorescent enhancement upon binding to ds-DNA. PicoGreen may bind via surface or groove interactions and can quantify as little as 25 pg/ml of ds-DNA in solution (Singer et al. 1997). This dye is 400-fold more sensitive than the Hoechst 33258 (Rengarajan et al. 2002).
The main purpose of this project was to develop a superior ultrasensitive ds-DNA histochemical staining technique. Additionally, we wanted to significantly improve the retention and location of intact ds-DNA, which was achieved by developing a novel histotechnological processing procedure [e.g., faster tissue sample acquisition (2 min or less), multiple fixation options, simultaneous microwave–vacuum oven processing, controlling humidity and static factors, thinner tissue sections, lower temperature tissue processing and slide drying, enzymatic antigen retrieval (AR), ultra-clean workstation, dim lights during PicoGreen staining, ultrapure water and reagents, and a computerized image-analysis system] and using lower melting point paraffin to reduce heating. We also determined which of the 15 fixatives worked best with our histological processing approach and DNA staining method. Together, these techniques result in a novel tissue-processing approach. Our group compared conventional processed tissue with tissue prepared by our new approach. Archival tissue was analyzed and the effects of delaying fixation. We found that extended prefixation times and improper processing severely compromise the final ds-DNA probe staining results (e.g., lower DNA content, incorrect cellular location, altered DNA structure, formation of denatured ss-DNA, and incorrect DNA ploidy analysis).
Our research resulted in the development of an extremely ultrasensitive assay for localizing and quantifying intact ds-DNA (i.e., right-handed B-DNA) in paraffin-embedded fixed tissue sections. This method also takes into account any possible problems with AR. Our overall histotechnological processing approach increased the retention of intact, undamaged ds-DNA content and maintained standard (i.e., B-DNA) and alternative (i.e., Z-DNA) nucleic acid structures. This study proves that it is feasible to simultaneously maintain excellent tissue morphology and the integrity of unaltered ds-DNA structures in histologically processed tissue.
Materials and Methods
Tissue Acquisition: Reduced Prefixation Time, Extended Postmortem Fixation, and 8-Year-Old Tissue Sections
Ten 18-month-old pigs were provided for the study by Max Insel Cohen, Animal Tissues for Research (Livingston, NJ). The procedures for removal of skin conformed to the practices established by the Institutional Animal Use Committee. Half of the tissues were processed by conventional histological procedures (Carson 1997; Gagna et al. 1997), whereas the other half of the tissues were processed by utilizing our novel processing method. To compare freshly fixed tissue to extended postmortem tissue fixation, six pig skin samples were processed. Half of these were stored for 24 hr in a cold room (+4C) before fixation, and the other half were fixed immediately (i.e., 2 min). Both were then processed using our novel histotechnological procedure. Additionally, in September 1999, skin tissue samples were obtained from 18-month-old pigs (Max Insel Cohen), immediately fixed for 72 hr in 10% unbuffered formalin, sectioned at 1.5-μm thick, and then stored at room temperature in a closed box. Eight years later they were stained with PicoGreen.
All skin samples were normal. Fixation of the skin was standardized for all 15 fixatives as to length of time, histological processing, thickness of tissue sections, microwave–vacuum oven usage, temperature, use of chemicals and reagents, reagent volume strength, and DNA probe staining. Microwave wattage and time need to be determined by the investigator due to temperature variations.
Skin was removed (<2 min prefixation time in a cold room, on ice), immediately fixed, and then processed. The skin was diced into pieces 1- to 3-mm3 thick, then placed into the fixative. Skin specimens consisted of the epidermis and the upper part of the dermis. The volume of fixative should be 35 times the volume of the tissue. To ensure consistent solution composition for all procedures, the Barnstead Easypure ultraviolet/ultrafiltration Type I reagent-grade water purification system (Barnstead; Dubuque, IA) was used to obtain ultrapure deionized water (DNase free, RNase free, and pyrogen free). Fresh and ultrapure chemicals, reagents, and biochemicals (e.g., molecular biology grade, Certified ACS Plus, GC Resolv, and OPTIMA; Fisher Scientific, Pittsburgh, PA) were also used in all the procedures unless otherwise specified. All glass and plastic containers were free of DNase, RNase, and pyrogen.
Tissue Fixatives
To determine which fixatives resulted in superior preservation of tissue-bound ds-DNA, skin was processed in 1 of 15 fixatives: Carnoy's solution (10 ml glacial acetic acid, 60 ml absolute ethanol, and 30 ml chloroform); Clarke's solution (20 ml glacial acetic acid and 60 ml absolute ethanol); zinc formalin fixative [10 ml 37% formaldehyde, electron microscopy (EM) grade, 90 ml water, and 1 g zinc sulfate]; Davidson's fixative [30 ml 95% ethanol, 30 ml 10% neutral-buffered formalin (NBF) (EM grade), 10 ml glacial acetic acid, and 30 ml water]; 10% ultrapure NBF (methanol-free EM grade buffered with acetate); 10% unbuffered formalin (histology grade); Streck Tissue Fixative (STF) (diazolidinyl urea, 2-bromo-2-nitropropane-1,3-diol, zinc sulfate, and sodium citrate) (Streck Laboratories; Omaha, NE); formalin–alcohol–acetic acid (FAA) [10 ml 39–40% formalin (EM grade), 90 ml 80% ethanol, and 5 ml glacial acetic acid]; Müller's fluid (2.5 g potassium dichromate, 1 g sodium sulfate, and 100 ml water); Flemming's strong solution(20 ml 2% osmium tetroxide, 75 ml 1% chromic acid, and 5 ml glacial acetic acid); Bouin's solution [75 ml picric acid, saturated aqueous, 25 ml 37–40% formaldehyde (histology grade), and 5 ml glacial acetic acid]; Zenker's solution (2.5 g potassium dichromate, 4.5 g mercuric chloride, 5 ml glacial acetic acid, 1 g sodium sulfate, and 100 ml water); 1% glutaraldehyde fixative [4 ml 50% glutaraldehyde (EM grade) and 100 ml 0.1 M phosphate buffer (pH 7.4)]; methacarn fixative (60 ml methyl alcohol, 30 ml chloroform, and 10 ml glacial acetic acid); and B-5 fixative (12 g mercuric chloride, 2.5 g sodium acetate, and 200 ml water). Chemicals used to make the fixatives were fresh and ultrapure unless otherwise specified (Polysciences; Warrington, PA or Fisher Scientific).
Simultaneous Microwave–Vacuum Oven Fixation
A microwave–vacuum oven (DFR-10 Bio Wave with ColdSpot, 3435 PELCO Vacuum Chamber; Ted Pella, Redding, CA) was used to shorten histological processing time from hours to minutes (Kok and Boon 1992; Jamur et al. 1995; Marani et al. 1996; Giberson and Demaree 2001) to reduce and/or prevent the loss of DNA content, location, and structure, which optimized PicoGreen probe staining. It has an adjustable power range (80–750 W) with continuous controlled power and a ColdSpot. Low-power microwave energy, together with the ColdSpot, can eliminate specimen heating (Giberson and Demaree 2001). Temperature is critical and should never exceed 35C–40C.
Ultrafast, primary, microwave–vacuum oven fixation of tissue was achieved at 35C ± 1C for 5 to 15 min (150 W) with all alcohol fixatives and Müller's solution, Flemming's solution, STF, Zenker's solution, B-5 fixative, and 1% glutaraldehyde. All other fixatives required a two-step microwave–vacuum oven fixation (45 min at 150 W and 15 min at 650 W; 35C) because of the formaldehyde content. Heating time was divided into two cycles with a 1-min interval between cycles to check on the fluid level in the jars. After heating, the jars were removed and allowed to cool for 10–15 min.
Histotechnological Processing of Tissues: Dehydration and Clearing
The microwave–vacuum oven (ColdSpot) was used (150 W) to dehydrate tissue in four alcohol solutions (viz., 70%, 95%, 100%, and 100% ethanol) with two solution changes at 40C for 7, 5, 5, and 5 min, respectively. Clearing tissue in chloroform, which causes less hardening to tissue than xylene, was also performed in the microwave–vacuum oven (650 W) with three changes at 40C for 10 min each (Kok and Boon 1992; Jamur et al. 1995).
Paraffin Embedding of Fixed Tissue
Skin was embedded in Paraplast X-tra Paraffin (52C–54C) (Electron Microscopy Sciences; Hatfield, PA) in a Fisher Isotemp 282A precision vacuum oven. Four changes of paraffin (300 ml) should be used at 53C ± 1C with an infiltration time of 3 hr. Overheating of paraffin during infiltration will harden the tissue samples and may denature the ds-DNA.
Tissue Sectioning, Deparaffinization, and Rehydration of Fixed Tissue Sections
All procedures were performed within a plastic–metal glove box with a HEPA filtration module to obtain pure air (Model 1689-003; Terra Universal, Anaheim, CA). This allowed for the control of humidity, temperature, and static factors to remove dust and other contaminants during tissue processing and PicoGreen staining. Skin was sectioned with a Carl Zeiss HM 325 rotary microtome (Thornwood, NY) in 1.5-, 5-, 7-, 10-, and 15-μm-thick specimens. To improve the quality of tissue sections, we used Accu-Edge blades (Ted Pella). The use of Paraplast X-tra was critical because it allowed for thinner sections (1.5 μm) and lower heating during tissue processing. Tissue sections were placed on Fisherbrand Superfrost Plus microscope slides. To avoid drying slides at 60C–80C, which has been shown to be harmful to many antigens (Jamur et al. 1995; Henwood 2005), slides were dried in the microwave–vacuum oven (35C) for 1–5 min using a microwave-safe slide rack (Simport Plastic; Beloeil, Canada) and then cooled.
Paraffin was removed from tissue sections at room temperature using chloroform (four changes, 2 min each) and rehydrated into water with ethanol [95% alcohol three changes, 30 sec each; 50% alcohol three changes, 30 sec each; 10% alcohol three changes, 30 sec each; water wash four changes, 30 sec each; and Tris-EDTA buffer, pH 7.5 (10 mM Tris-HCl and 1 mM EDTA) (TE) five changes, 30 sec each]. The use of PicoGreen cannot be performed in tissue sections dehydrated in alcohol (data not shown). Some sections were stained with hematoxylin and eosin (H/E) as control slides.
AR for Enhancement of PicoGreen Probe Histochemical Staining
To allow PicoGreen to react with all binding sites of the ds-DNA, it may be necessary to use AR (Huang et al. 1976; Roth et al. 2000; Shi et al. 2000). AR will differ depending on tissue, fixative, fixation times, and processing. Enzymatic AR was used instead of high-temperature AR, which requires 100C and would denature ds-DNA and alter DNA structure (data not shown). AR was performed either before or after microwave–vacuum oven fixation. Enzymatic AR was achieved by placing tissue sections into a 10-slide Coplin glass-staining jar. Tissue sections were prewarmed (37C) in TE buffer and then placed into 0.1% trypsin in 0.1% calcium chloride (37C) [400 ml water, and 5% calcium chloride (8 ml), trypsin (100 mg) (Fisher Scientific), pH 7.6] (Huang et al. 1976). They were incubated in a darkened moisture chamber (Evergreen Scientific; Los Angeles, CA), within the Terra Universal glove box for 2–40 min, then washed three times for 4 min each in cold water, followed by five washes for 2 min each in TE buffer at room temperature.
PicoGreen Histochemical Staining of Fixed Tissue Sections
PicoGreen ds-DNA fluorescent stain (Molecular Probes; Eugene, OR) is an ultrasensitive nucleic acid probe. PicoGreen dye was diluted (1:200) in TE buffer at room temperature. The working solution of PicoGreen reagent is made by diluting the concentrated dimethylsulfoxide solution in TE buffer. All dilutions must be made in dark-colored plastic polypropylene tubes just before use (30 min or less) and carefully vortexed. PicoGreen is susceptible to photodegradation from room light so it must be handled only with the lights dimmed. Dilution of PicoGreen for different types of tissues processed with different fixatives may have to be determined independently, i.e., a dilution series to assess the optimal concentration for PicoGreen.
Using a Brinkmann Eppendorf single-channel micropipettor (Fisher), 2000 μl (low-retention Fisherbrand tips) of PicoGreen were placed on each tissue slide, then incubated in a moisture chamber (Evergreen Scientific) housed in the Terra Universal glove box (slides should not touch one another) in total darkness at 25C for 5 to 15 min. Our results indicated that PicoGreen staining cannot be performed with the microwave–vacuum oven because the probe absorbs microwave energy and the PicoGreen breaks down (data not shown). We used a liquid blocker pen (Ted Pella) to draw a circle around the tissue on the glass slide to hold the PicoGreen solution. With the lights off and at room temperature, using the glove box, PicoGreen-treated tissue sections were then washed five times for 3 min each in TE buffer (4C). Tissue sections were always completely saturated. This was followed by three 4-min washes in water. Slides were then mounted (aqueous mounting medium) (Aqua-Poly/Mount; Polysciences) and coverslipped in a darkened environment. The Aqua-Poly/Mount (non-fluorescent) is stored at 4C but should be left at room temperature for 4 hr before use. Stained slides were left for 30 min at room temperature in absolute darkness before quantifying PicoGreen. Two negative controls were used: omission of PicoGreen substituted with TE and prior use of chemical or enzymatic extraction of DNA (Gagna et al. 1997). A positive control was also used (Gagna et al. 1997).
Drug and Fluorescent Dye Inhibition Experimentation
Tissue sections were pretreated with drugs or dyes to test for their effectiveness in inhibiting the binding of PicoGreen and as additional controls. All dilutions of drugs or dyes should be made using TE buffer. The following five drugs and dyes were applied individually as pretreatments before using PicoGreen (Haugland 2002). The intercalating agent actinomycin D [guanine–cytosine (G–C) specific (0.1 to 0.4 μg/ml)]; the thymidine analog 5-bromodeoxyuridine (25–200 μg/ml); ethidium bromide (ds-DNA fluorescent intercalator) (0.5 to 10 mg/ml); the antibiotic mitramycin (G–C-specific non-intercalator) (10–1000 nM); and Hoechst 33258 [adenine– thymine (A–T) specific] (fluorescent dye, minor-groove binding) (0.1–10μg/ml). After inhibition procedures, tissue sections were washed five times for 4 min each with TE buffer at room temperature and then stained with PicoGreen.
Nuclease Digestion of Skin Tissue Sections
The following pretreatments were used as controls and to eliminate ss-DNA and RNA. As a control, sections were incubated in the dark (moisture-chamber glove box) with DNase I, RNase Free (Boehringer Mannheim; Indianapolis, IN). Full DNase I digestion consisted of 0.5 mg/ml of DNase I in 50 mM Tris, pH 7.5, 10 mM MgSO4, 0.1 mM dithiothreitol, and 50 μg/ml bovine serum albumin for 10 min to 3 hr at 20C. The nicking digestion solution contained 5–10 ng/ml of DNase I (Gagna et al. 1997).
Fluorescence due to PicoGreen binding to RNA at high concentrations can be eliminated by treating the sample with DNase-free RNase. Therefore, to remove RNA from tissues, RNase ONE (DNase-free) solution (Promega; Madison, WI) was used (0.5 mg/ml) in PBS (pH 7.3, 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4·7H2O, and 1.4 mM KH2PO4) for 1–9 hr at 37C in a moisture chamber in the dark (Gagna et al. 1997).
To ensure that fluorescence of the entire tissue sample is due to ds-DNA, all ss-nucleic acids were eliminated. ss-DNA and ss-RNA were removed from fixed tissue by using S1 nuclease (Vogt 1980), RNase A/RNase T1 (Sambrook et al. 1989), or mung bean nuclease (Kowalski et al. 1976) (Promega). After enzyme digestion, tissues were washed four times for 5 min each with water at room temperature, then four times for 2 min each with TE buffer at room temperature, and then stained with PicoGreen.
PicoGreen–Nucleic Acid Histochemical Competition Procedures
Validity of the PicoGreen probe-staining specificity for the fixed tissue-bound ds-DNA was determined by using purified low and high molecular weight nucleic acids. All dilutions of nucleic acids were made with TE buffer. Many antigen competitors were used such as ds-B-DNA, ds-Z-DNA, ss-DNA, ss- and ds-RNA, triplex-DNA, quadruplex-DNA, DNA–RNA hybrids, RNA–RNA hybrids, poly[d(G–me5C)], poly[d(G–C)], poly(G–T)–poly(A–C), or poly(A–G)–poly(C–T) (native nucleic acids and synthetic polynucleotides) (Pharmacia Biotech; Piscataway, NJ) (Gagna et al. 1997). Before PicoGreen staining of fixed tissue sections, the probe (0.5–2000 μg/ml) was preincubated with antigen competitors of low or high molecular weight (Gagna et al. 1997). After PicoGreen staining, tissue sections were washed five times for 3 min each in TE buffer at room temperature in the dark.
Computer-image Acquisition and Analysis: PicoGreen Fluorescent Histochemical Probe
This novel DNA staining technique allows for the image cytometric measurement of fluorescence distribution and intensity of fixed tissue-bound ds-DNA content. Quantitative PicoGreen binding data were expressed in mean optical density (MOD) units (Gagna et al. 1997). Microspectrofluorometric measurements of PicoGreen were performed using the Leitz DM-RB compound microscope (Postfach, Germany) (Poulin et al. 1994; Singer et al. 1997; Kuo et al. 1998). Fluotar objectives were used (×200–×1000). Tissue sections were located under transmitted light and then switched to UV illumination to quantify PicoGreen binding. It is imperative to immediately examine the preparations because fluorescence fades rapidly and/or dissociates from PicoGreen-stained cells. Images were obtained with an Optronics DEI-750 CE digital camera (Optronics; Bolton, MA) and an imaging-analysis system (Optronics S60671) in a totally darkened room. Images were then sent into Image Pro-Plus 4.0 (Media Cybernetics; Silver Spring, MD). A Dell OptiPlex GX1 and Windows NT (Workstation 4.0) were used to perform all quantitative procedures (Kuo et al. 1998). Corrections of non-uniformity arc lamp epillumination over the field of view were performed for proper quantitative measurement (Poulin et al. 1994; Singer et al. 1997; Kuo et al. 1998). PicoGreen MOD measurements were made at room temperature, and tissue samples were excited at a wavelength of 480 (±10) nm and emission measurements performed at 520 (±12.5) nm. Microscopic fluorescence was calculated for each tissue-section image. Readings were taken after 15 sec or less. Blank values and background fluorescence were subtracted from the raw data. Use of negative and positive controls allowed for comparable quantification of the results (Poulin et al. 1994; Kuo et al. 1998). Triplicate slides with continuous serial tissue sections were simultaneously processed. For positive controls, the Feulgen reaction for total DNA content and anti-B-DNA antibody probe for ds-B-DNA content were used (Gagna et al. 1997).
Z-DNA Immunohistochemistry
To characterize the effects of fixatives and the ability of our histological processing procedure to maintain DNA structure, we stained tissue sections with anti-Z-DNA antibodies (Gagna et al. 1997). The anti-Z-DNA polyclonal antibody (PAb)(Z-4255) and anti-Z-DNA monoclonal antibody (MAb) (Z-44) were used to stain tissues, half of which were processed conventionally (Carson 1997; Gagna et al. 1997) and half that were processed using our novel procedure. Staining of the experimental and control tissues [anti-DNA IgG PAb (10–100 μg/ml) or anti-DNA IgG MAb (1–10μg/ml)] was performed using the avidin–biotin peroxidase complex method (Vector Laboratories; Burlingame, CA) (Gagna et al. 1997).
Enhancement of Z-DNA Immunoreactivity With 45% Acetic Acid
To reveal or to enhance Z-DNA immunoreactivity, tissue sections were exposed to 45% glacial acetic acid (ultrapure grade; Gallard-Schlesinger, Carle Place, NY) with three changes for 5 min each at room temperature in the dark (moisture-chamber glove box) and then rinsed four times in TE buffer for 5 min each, prior to anti-Z-DNA antibody staining (Gagna et al. 1997). The 45% acetic acid should be considered an AR procedure for Z-DNA.
Computerized Image Acquisition and Analysis: Anti-Z-DNA Immunohistochemistry
Non-fluorescent image acquisition and analysis for Z-DNA immunohistochemistry (MOD) [3,3′-diaminobenzidine (DAB)] was performed as described by Gagna et al. (1997) using the Leitz DM-RB microscope. A quantitative measure of reactive products was generated in the Quantiment Performer Interactive Programming System in the Quantiment 500+ image analysis system (Leitz; Cambridge, UK). Positive and negative controls were used (Gagna et al. 1997).
Statistical Analysis
Results from serial tissue sections of 10 different specimens processed in 15 fixatives using PicoGreen and anti-Z-DNA antibodies, anti-B-DNA antibody staining, and Feulgen reaction (and controls) were reproduced. Use of a single PicoGreen probe and Z-DNA MAb and PAb was repeated six times (n=6). Results were statistically analyzed using one-way ANOVA program (Sigma Stat; Jandel, IL). Data were expressed as mean ± SD, and the statistical significance of each experiment was p<0.05.
Results
Tissue Fixatives and PicoGreen ds-DNA Probe Histochemistry
Tissue fixed in Carnoy's solution (Figure 1), methacarn, and Clarke's solution, then processed by our novel histotechnological procedure and DNA histochemical staining technique (Figure 2), yielded the highest quantitative (MOD) PicoGreen staining results (Figure 3). These fixatives also allowed for distinct tissue morphology and nuclear details (Figure 1 and Figure 2). FAA, STF (Figure 3), Davidson's, zinc formalin, 1% glutaraldehyde (Figure 4), and 10% ultrapure NBF (Figure 5) fixatives produced the next highest group of PicoGreen probe-stained tissue sections. Fixatives that generated the lowest PicoGreen staining were 10% unbuffered formalin (Figure 5), Bouin's solution, Zenker's solution, Flemming's strong solution, Müller's solution, and B-5 fixative (Figure 6). However, they produced excellent tissue fixation. Even when Zenker's solution was posttreated to wash out the chromate, PicoGreen staining was not significantly improved (Figure 6). The same results occurred with Bouin's solution when washed to remove picric acid. When tissues processed in B-5 fixative, Flemming's strong solution, and Müller's fluid were washed to remove mercuric chloride, chromic acid, and potassium dichromate, respectively, MOD binding values actually declined (Figure 6). Tissue fixed in Carnoy's solution, methacarn, and Clarke's solution and then processed with AR prior to PicoGreen staining generated higher MOD binding values (Figure 3). With these three fixatives, AR increased PicoGreen binding values by an average of 34%, 29%, and 39%, respectively. Whereas alcohol fixatives usually shrink and harden tissue, our histotechnological processing approach produced tissues that were softer, regardless of fixative. This approach also produced superior H/E-stained tissue sections (Figure 1). Control values for PicoGreen were 59 ± 21 MOD binding units.
Histotechnological Processing of Tissue Samples: Thickness of Tissue Sections
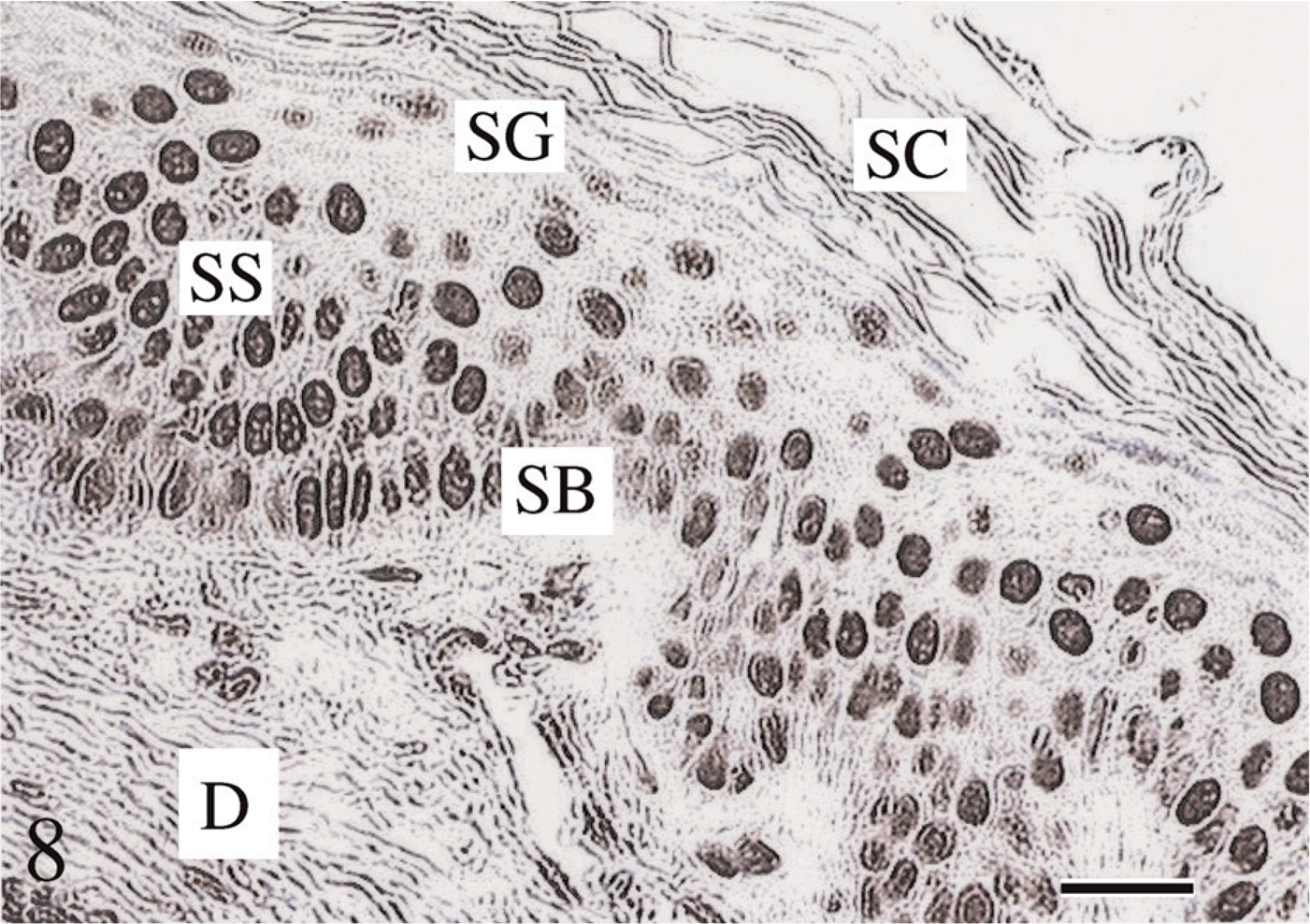
Tissue sections 1.5-μm thick offered the best demonstration of tissue morphology (Figure 1) and the highest PicoGreen and Z-DNA immunoreactivity MOD binding values (Figure 2–Figure 11). Five- to 7-μm-thick tissue sections, and especially 10- to 15-μm-thick tissues, were of inferior quality and produced lower binding values (14% and 17% decrease, respectively). The novel histotechnological processing procedure (i.e., 1.5-μm-thick sections) allowed for superior morphology of tissue sections compared with conventional processing procedures (i.e., 1.5-μm-thick sections) (Figure 1, Figure 2, and Figure 8).
Novel Histotechnological Processing Procedure Compared With Conventional Tissue Processing Greatly Enhances PicoGreen Histochemistry
Our histotechnological processing procedure (without AR), compared with the conventional tissue processing procedure (without AR), generated a 111% increase in PicoGreen MOD binding values for tissue fixed in Carnoy's solution (Figure 2 and Figure 7). Our new tissue-processing approach (with AR) produced a 124% increase in PicoGreen probe histochemical staining, compared with conventional processing (with AR) for tissue fixed in Carnoy's solution (Figure 3 and Figure 7). PicoGreen probe binding increased 198% when we used our histotechnological processing approach with AR, compared with conventional tissue processing without AR. Use of paraffin with a lower melting temperature (i.e., 53C–55C instead of 62C) reduced exposure of ds-DNA to heat and increased PicoGreen binding by 17% and Z-DNA immunoreactivity by 47%. Optimal time for tissue incubation (i.e., staining) of PicoGreen was 12 min. Overincubation (i.e., 180 min and, especially, 280 min) significantly enhanced background noise. Underincubation (i.e., 1 and 2 min) resulted in no histochemical fluorescence or very weak (i.e., 3–4 min) fluorescence (data not shown). Not using dimmed light for the PicoGreen staining protocol produced lower binding values (39% decrease). PicoGreen staining in full daylight resulted in an 89% decrease in MOD binding values.
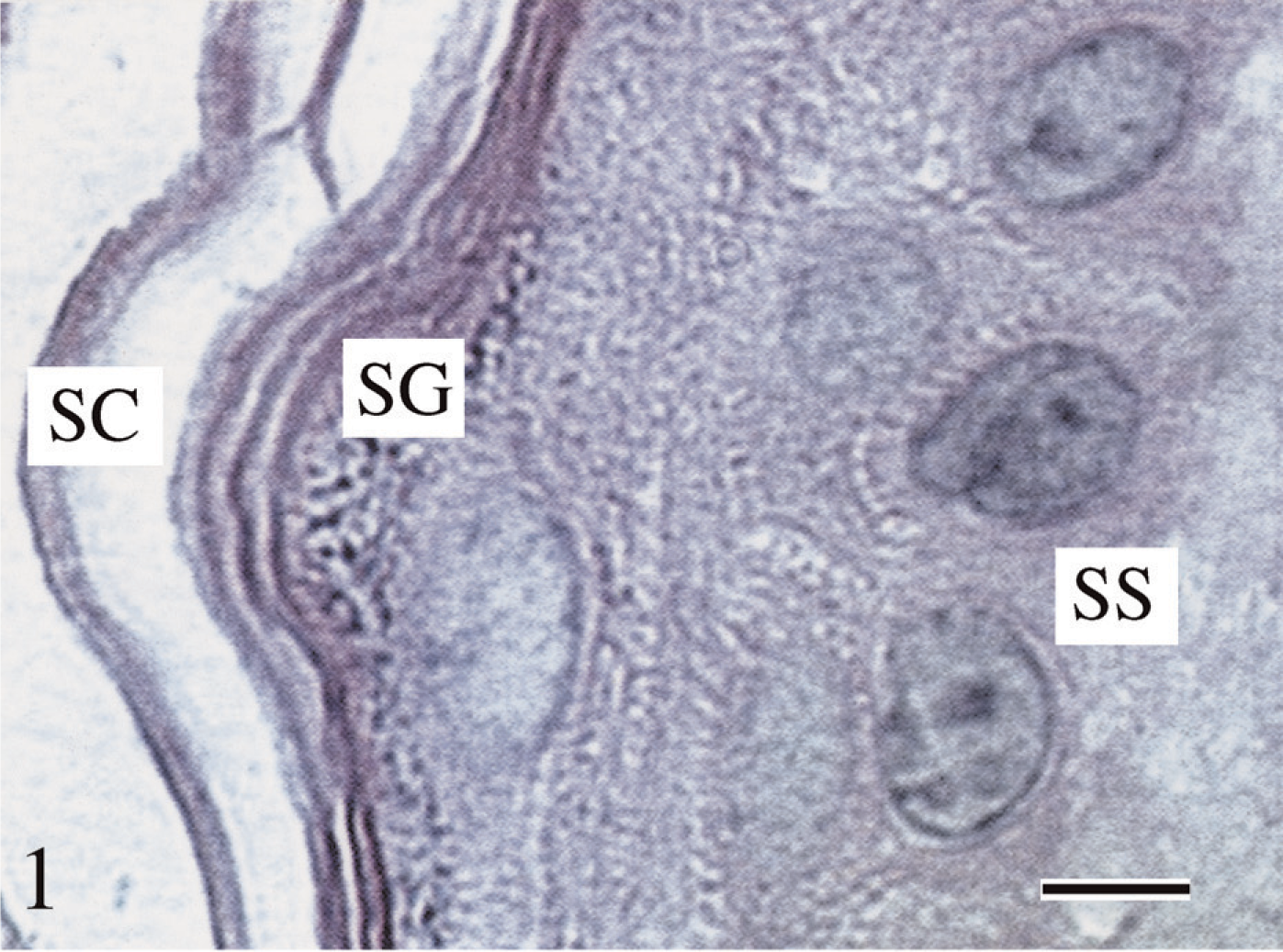
Hematoxylin/eosin (H/E) stained tissue section. Tissue was fixed in Carnoy's solution, processed using our histotechnological procedure, and then stained with H/E. The 1.5-μm-thick tissue sections resulted in the best demonstration of overall tissue morphology and, especially, nuclear preservation. The novel tissue processing procedure enhances the morphology of all the epidermal strata, viz., stratum corneum (SC), stratum granulosum (SG), and stratum spinosum (SS). Bar = ∼5 μm.

Fluorescent staining of a fixed tissue section using the PicoGreen probe. Tissue was fixed in Carnoy's solution, processed using our novel histotechnological procedure, and then histochemically stained with the PicoGreen probe. Fluorescent staining reveals intact undamaged ds-DNA within the nuclei of the epidermis, viz., SC, SG, and SS. Tissue sections were treated with antigen retrieval (AR). Tissue sections are 1.5 μm thick. Bar = ∼2.5 μm.
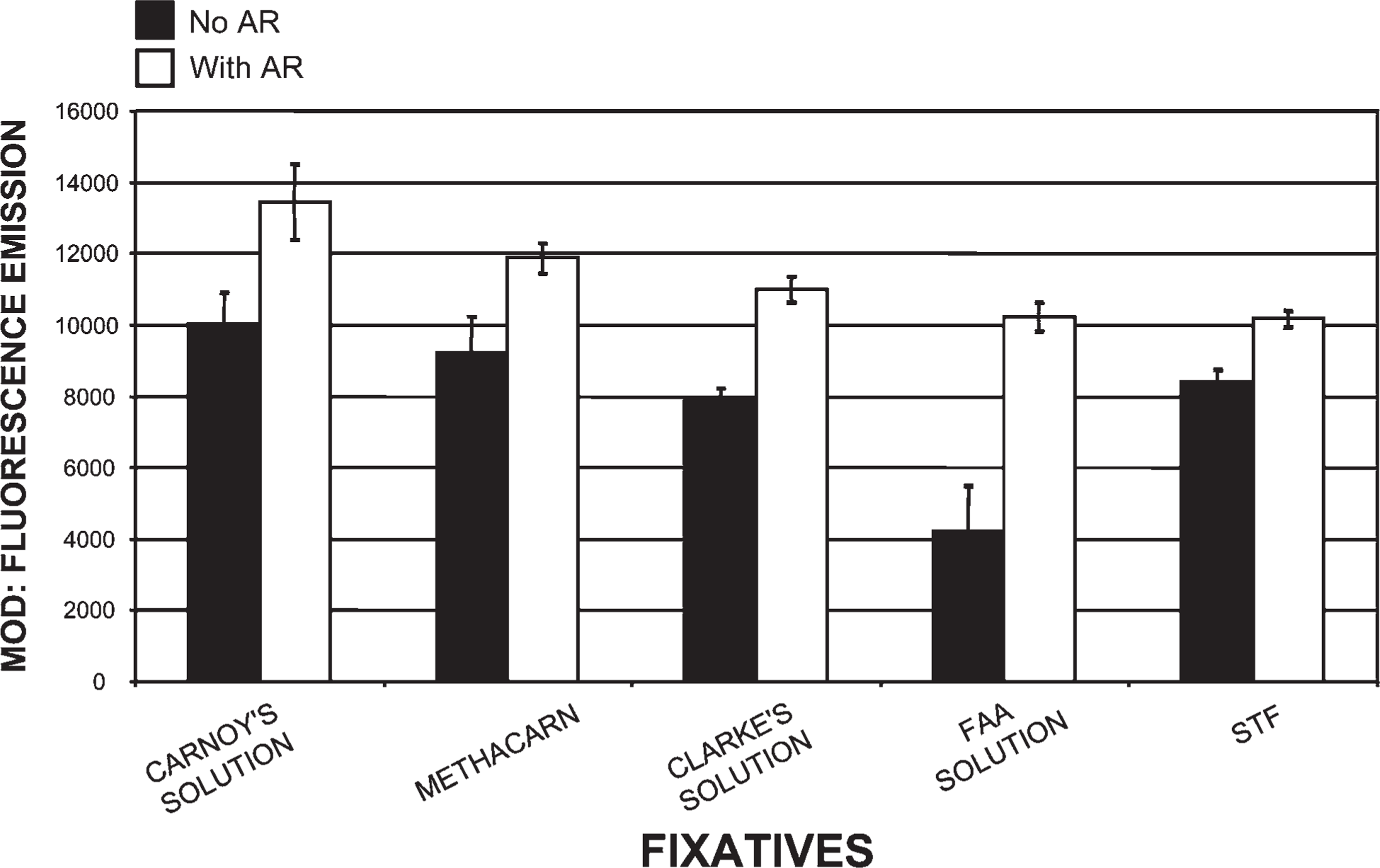
Comparison of tissues processed in five different fixatives using our histotechnological processing procedure and then stained with PicoGreen. This figure compares Carnoy's solution, which allowed for the highest fluorescent mean optical density (MOD) binding values of all 15 fixatives to four other fixatives: methacarn, Clarke's solution, formalin–alcohol–acetic acid (FAA) solution, and Streck Tissue Fixative (STF). This figure also reveals how AR increased PicoGreen binding. FAA was the only fixative in this group that contained formaldehyde, so it required the most AR. Of all five fixatives, STF required the least amount of AR. Tissue sections are 1.5-μm thick.
AR Enhances PicoGreen Histochemistry Depending on Fixative
Enzymatic AR was used to increase exposure of binding sites of the fixed tissue-bound ds-DNA (Poulin et al. 1994; Roth et al. 2000). Alcohol-based fixatives required the least amount of AR (i.e., 5–10 min) and produced a 34% increase in MOD binding values (Figure 3, Figure 4, and Figure 7). Conversely, cross-linking fixatives required the most AR (i.e., 15–50 min) (Figure 3 and Figure 4–Figure 7). AR improved PicoGreen quantitative values for the cross-linking fixatives (i.e., 1% glutaraldehyde, 10% ultrapure NBF, and 10% unbuffered formalin), yielding a 241% increase in binding (Figure 4 and Figure 5). FAA and Davidson's solutions, fixatives that contain both precipitating (alcohol) and cross-linking (aldehyde) elements, increased PicoGreen binding by 150% after AR (i.e., 10–20 min). All other fixatives (viz., Bouin's, Zenker's, Flemming's, Müller's, and B-5 solutions) exposed to AR generated an average increase of 103% for PicoGreen staining. However, the average of these five fixatives with AR yielded a 94% decrease in PicoGreen binding compared with Carnoy's solution with AR. Enzymatic pre-treatment, especially of alcohol-fixed tissues, prior to microwave–vacuum oven fixation caused tissue disintegration and decline in ds-DNA (73% decrease) and Z-DNA (100% decrease).
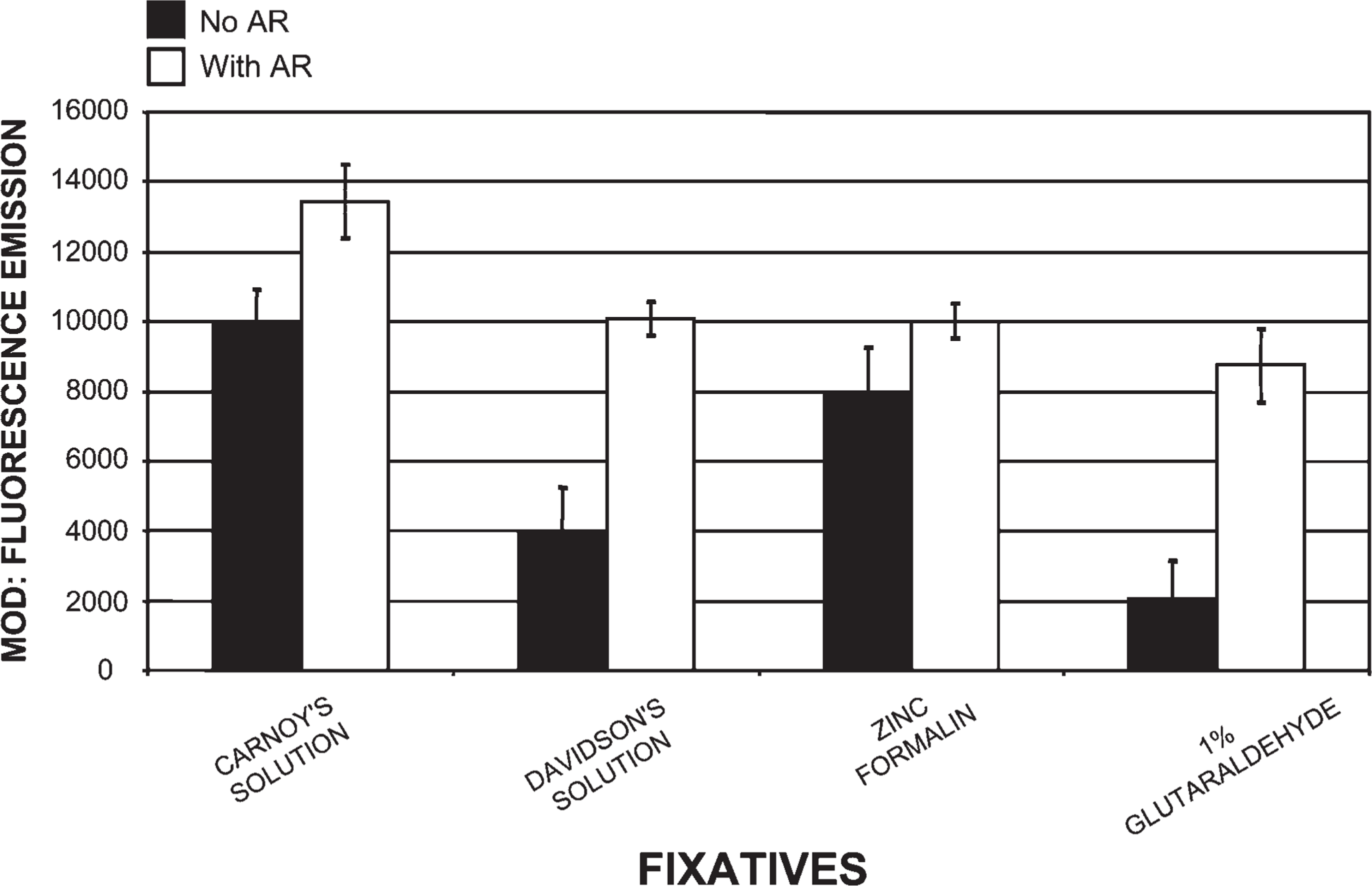
Comparison of tissues processed in four different fixatives using our histotechnological processing procedure and then stained with PicoGreen. This figure compares Carnoy's solution, which allowed for the highest fluorescent MOD PicoGreen binding values of all of our fixatives, to three other fixatives: Davidson's solution, zinc formalin, and 1% glutaraldehyde. This figure also reveals how AR increased PicoGreen MOD binding. Davidson's fixative and 1% glutaraldehyde required the most AR, whereas Carnoy's solution and zinc formalin required the least AR. Tissue sections are 1.5-μm thick.
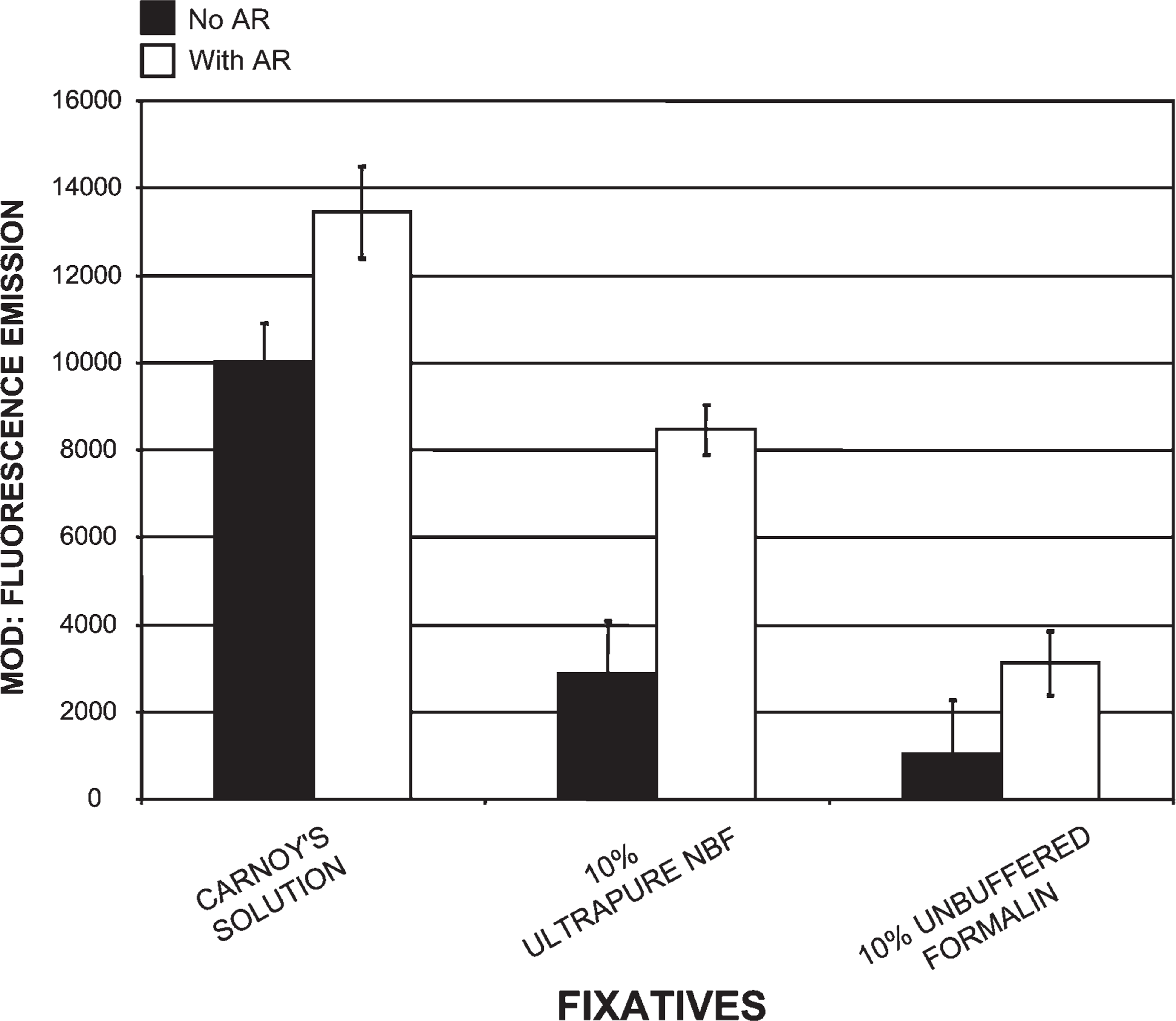
Comparison of tissues processed in three different fixatives using our histotechnological processing procedure and then stained with PicoGreen. This figure compares Carnoy's solution, which produced the highest MOD binding data, to two other fixatives: 10% ultrapure neutral-buffered formalin (NBF) and 10% unbuffered formalin. This figure also reveals how AR increased PicoGreen MOD binding. The 10% unbuffered formalin fixative resulted in the lowest PicoGreen staining of all three fixatives and required the most AR. Tissue sections are 1.5-μm thick.
Eight-Year-Old Tissue Sections and Delayed Fixation Compared With Our Novel Histotechnological Processing Procedure
Paraffin-embedded tissues were immediately fixed for 72 hr in 10% unbuffered formalin, sectioned (1.5 μm), and placed on slides in September 1999 and stored for 8 years at room temperature. They were then stained with PicoGreen, producing a 52% decrease in binding compared with PicoGreen staining, utilizing our histotechnological processing technique. A 31% decline in PicoGreen MOD binding value was calculated with postmortem tissue that was stored in a cold room for 24 hr before fixation and then processed using our new procedure.
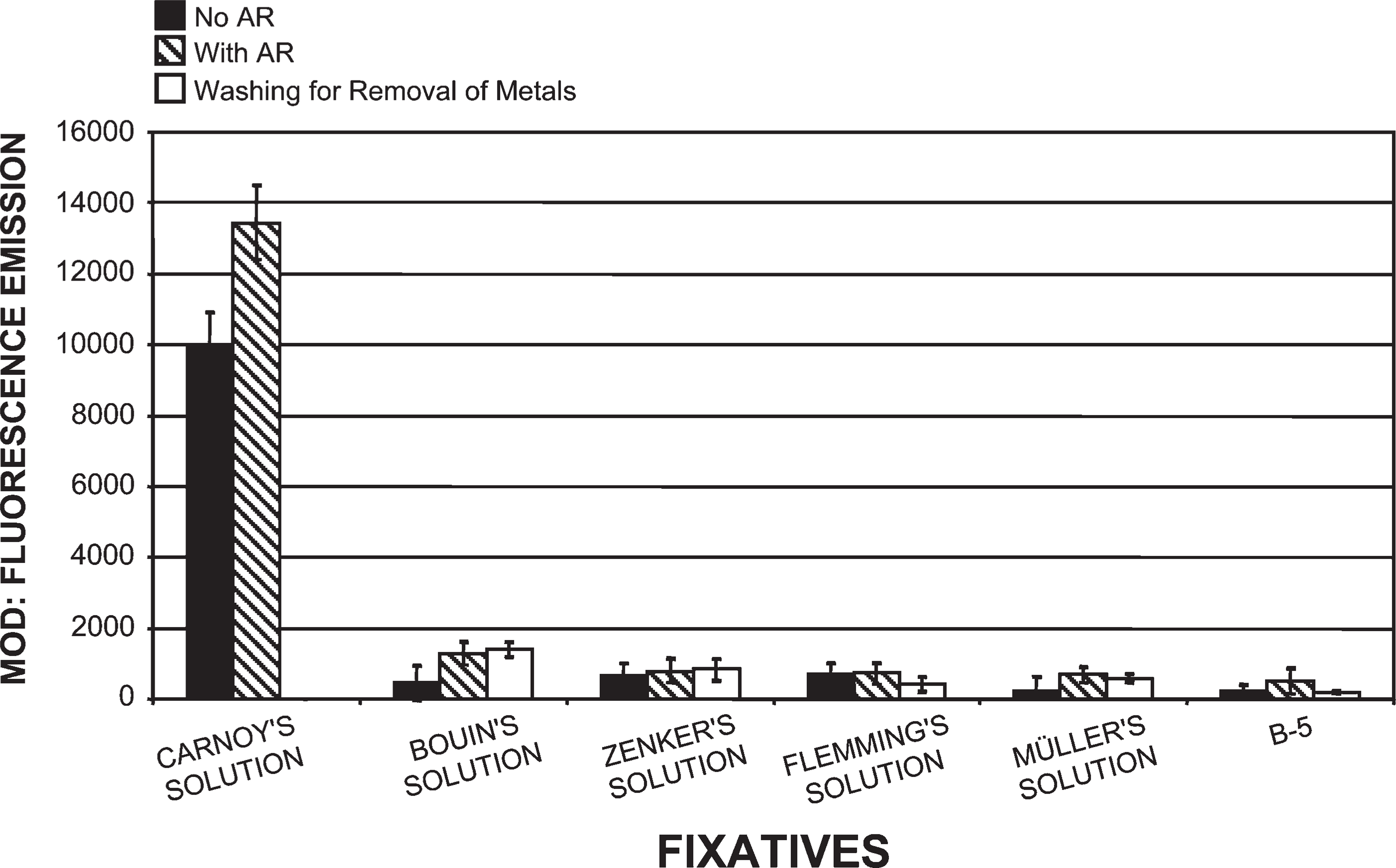
Comparison of tissues (1.5-μm thick) processed in six different fixatives using our histotechnological processing procedure and then stained with PicoGreen. This figure compares Carnoy's solution, which allowed for the highest fluorescent MOD binding values to five other fixatives: Bouin's solution, Zenker's solution, Flemming's strong solution, Müller's solution, and B-5 fixative. These five fixatives resulted in the lowest PicoGreen MOD values of all 15 fixatives. Removal of metals from fixatives by washing in water or chemicals actually resulted in a loss of PicoGreen binding with Flemming's strong solution, Müller's solution, and B-5 fixative. AR caused a slight increase in MOD binding values. Tissue sections are 1.5-μm thick.
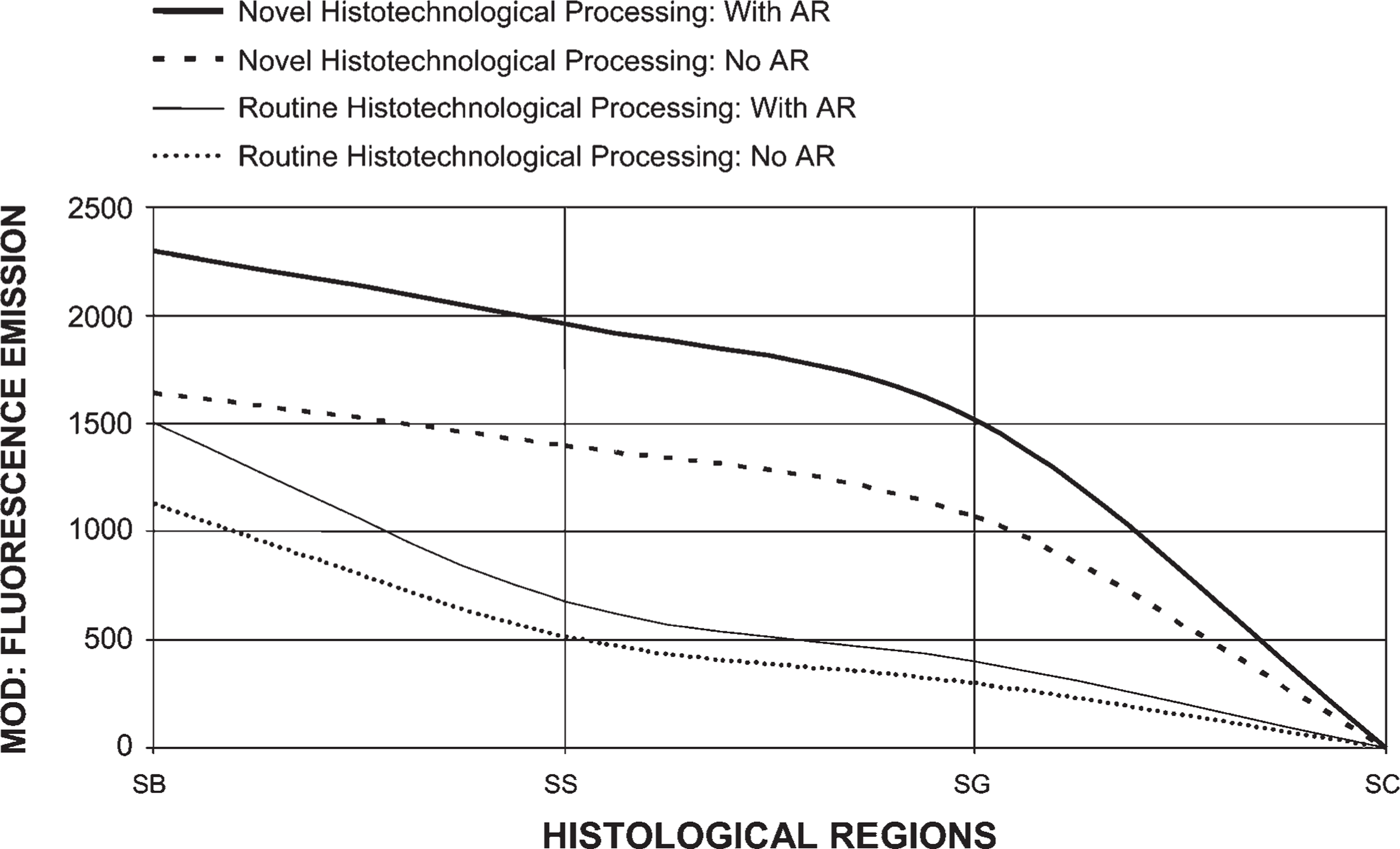
Comparison of PicoGreen staining with tissue processed by conventional procedures vs our histotechnological processing approach (with and without AR). Tissues were fixed in Carnoy's solution, which allowed for the highest fluorescent MOD binding to double-stranded (ds)-DNA. PicoGreen binding increased 198% when we used our histotechnological processing approach with AR, compared with conventional processing without AR. This figure reveals how the quantification (MOD) of ds-DNA can be improved by using our novel histotechnological processing procedure and histochemical PicoGreen staining technique. The epidermis is divided into four parts, viz., stratum basale (SB), SS, SG, and SC. Tissue sections are 1.5-μm thick.
Pretreatment With Drugs or Fluorescent Dyes Inhibited PicoGreen Probe Histochemistry
The use of drug or dye pretreatments before applying PicoGreen further supports the ultrasensitive nature of this probe. Pretreatments either reduced or completely inhibited PicoGreen binding. These results suggest that the drugs or dyes are blocking the ds-DNA binding sites and/or causing changes in the structure of right-handed ds-B-DNA to a non-B-DNA form such as Z-DNA. This, in turn, prevents PicoGreen from binding to the appropriate tissue-bound ds-DNA. Pre-treatments were influenced by concentration, purity, temperature, AR, and time of exposure.
Nuclease Digestion Pretreatment Eliminated PicoGreen Probe Histochemistry
Full DNase I pretreatment completely eliminated PicoGreen fluorescence, and nicking concentrations of DNase I reduced the binding reaction by 57%. Mung bean nuclease and RNase A/RNase T1 and S1 nuclease pretreatments had no effect on the quantifying of ds-DNA using PicoGreen. RNase ONE had no effect on PicoGreen tissue staining. This indicates that PicoGreen is binding to intact right-handed ds-B-DNA and not to ss-DNA or RNA. Pretreatments were dependent on concentration, exposure, purity, AR, and temperature.
DNA Competition Procedures Prevented PicoGreen Probe Histochemistry
PicoGreen staining of fixed tissue sections was inhibited after prior absorption of the PicoGreen reagent with ds-B-DNA. No reduction of fluorescent histochemistry occurred after tissue incubation of the PicoGreen reagent with any of the other nucleic acid competitors. No differences were observed between natural and synthetic DNAs and high and low molecular weight DNAs. These results indicate the specificity of PicoGreen toward fixed tissue-bound ds-DNA.
Z-DNA immunohistochemistry. This picture reveals anti-Z-DNA immunoreactivity (MOD): DAB (Z-4255 PAb) within the epidermis, viz., SC, SG, SS, and SB, as well as the dermis (D). Tissue was pretreated with 45% acetic acid (i.e., antigen retrieval). Z-DNA (DAB) was quantified as MOD units. Tissue sections are 1.5-μm thick. Bar = ∼30 μm.
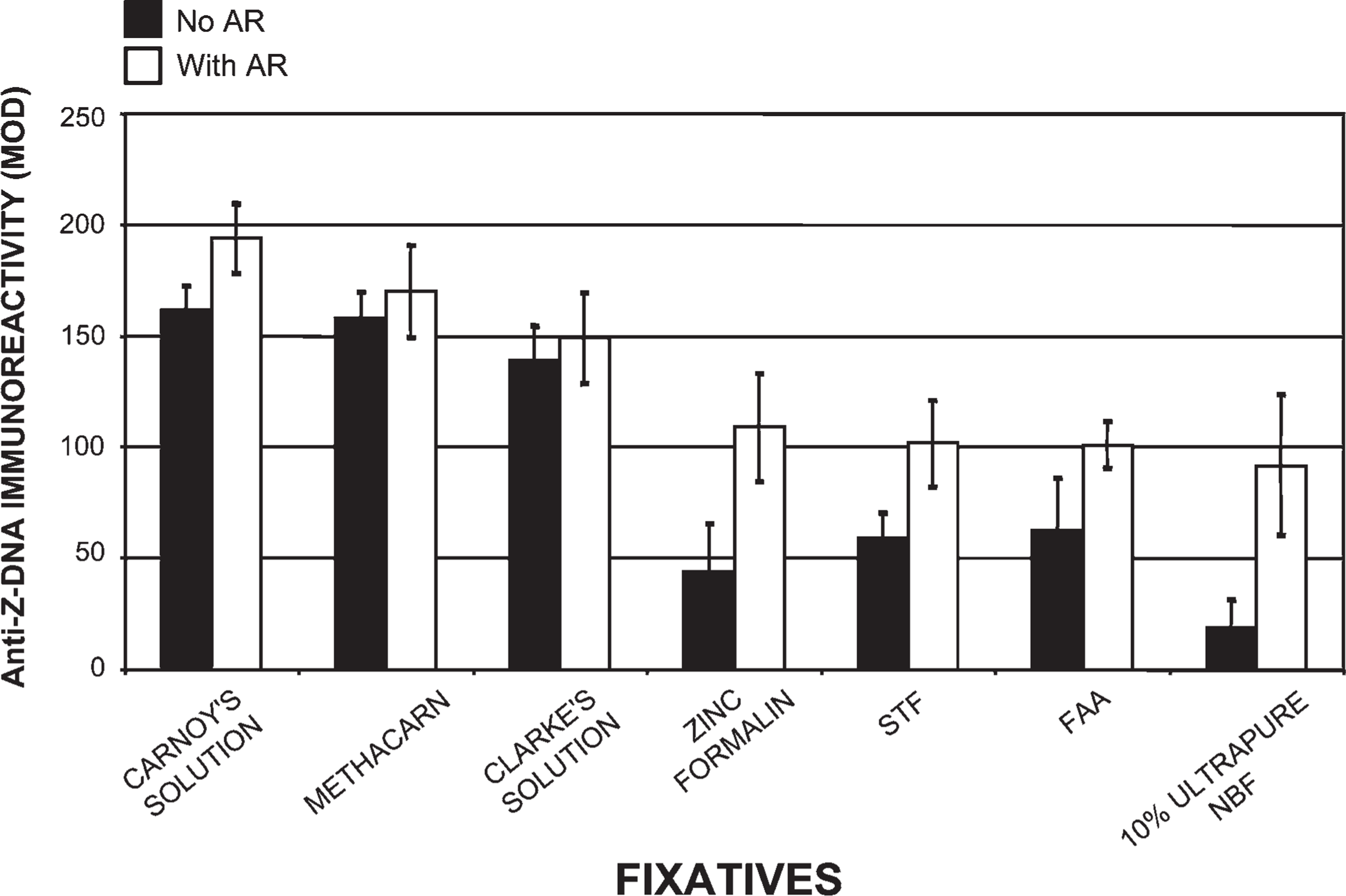
Effects of fixatives on Z-DNA immunoreactivity. Our novel histotechnological processing procedure was used for testing the effect of fixatives on Z-DNA immunohistochemistry (DAB). All fixatives were compared with Carnoy's solution, and data are expressed as MOD units. This figure exhibits Z-DNA immunoreactivity (Z-4255 PAb) before and after 45% glacial acetic acid treatments (i.e., AR). Seven fixatives were used: Carnoy's solution, methacarn, Clarke's solution, zinc formalin, STF, FAA, and 10% ultrapure NBF. Forty five percent acetic acid pretreatments enhanced the manifestation of Z-DNA MOD (DAB) binding values in all seven fixatives. Carnoy's solution, methacarn, and Clarke's solution resulted in the highest levels of Z-DNA immunoreactivity (MOD), whereas the last four fixatives resulted in the lowest anti-Z-DNA antibody binding, even with 45% acetic acid pretreatments. Zinc formalin and 10% ultrapure NBF required the most 45% acetic acid pretreatments. All tissue sections are 1.5-μm thick.
Z-DNA Immunoreactivity Was Enhanced by Our Novel Histotechnological Processing Procedure and Antigen-retrieving 45% Acetic Acid Pretreatment
Use of our histotechnological processing procedure greatly enhanced Z-DNA MOD (DAB) immunoreactivity (Figure 8–Figure 11). Compared with tissues processed by the conventional histological processing procedure (without 45% acetic acid) and fixed in Carnoy's solution, our processing approach (without 45% acetic acid) increased Z-DNA immunoreactivity by 309% (Figure 11). The use of 45% acetic acid pretreatments (AR) (i.e., prior to antibody staining) along with our histological processing procedure compared with conventional tissue processing (with 45% acetic acid) generated a 224% increase in Z-DNA (Figure 11). Our novel processing procedure (with 45% acetic acid) yielded a 717% increase in Z-DNA compared with conventional histotechnological processing (without 45% acetic acid) for tissues fixed in Carnoy's solution (Figure 11). Alcohol fixatives produced superior results compared with the aldehyde fixatives (Figure 9 and Figure 10). Even with 45% acetic acid pretreatments, tissue fixed in 10% ultrapure NBF produced lower Z-DNA MOD antibody staining than tissue processed in alcohol fixatives (without 45% acetic acid) (Figure 9). Tissue fixed in Carnoy's solution, methacarn, and Clarke's solution yielded the highest Z-DNA immunoreactivity (Figure 9). Zinc formalin, STF, FAA, and 10% ultrapure NBF fixatives produced lower Z-DNA antibody binding (Figure 9). Use of 10% unbuffered formalin, Bouin's, Flemming's, Müller's, Zenker's, and B-5 solutions (without AR) generated the lowest Z-DNA immunoreactivity close to background levels (Figure 10). With these fixatives, 45% acetic acid just barely initiated Z-DNA immunoreactivity. Eight-year-old paraffin-embedded tissue sections and tissues where fixation was delayed for 24 hr produced an 89% and 76% decline in Z-DNA, respectively, compared with tissue processed by our novel procedure. Full and nicking DNase I pretreatments prevented Z-DNA immunoreactivity. Our processing procedure is even more critical for obtaining accurate data when examining alternative DNA structures such as Z-DNA, vs B-DNA (Figure 8–Figure 11). Both Z-4255 PAb and Z-44 MAb produced similar results. Control values were 5±4 MOD binding units.
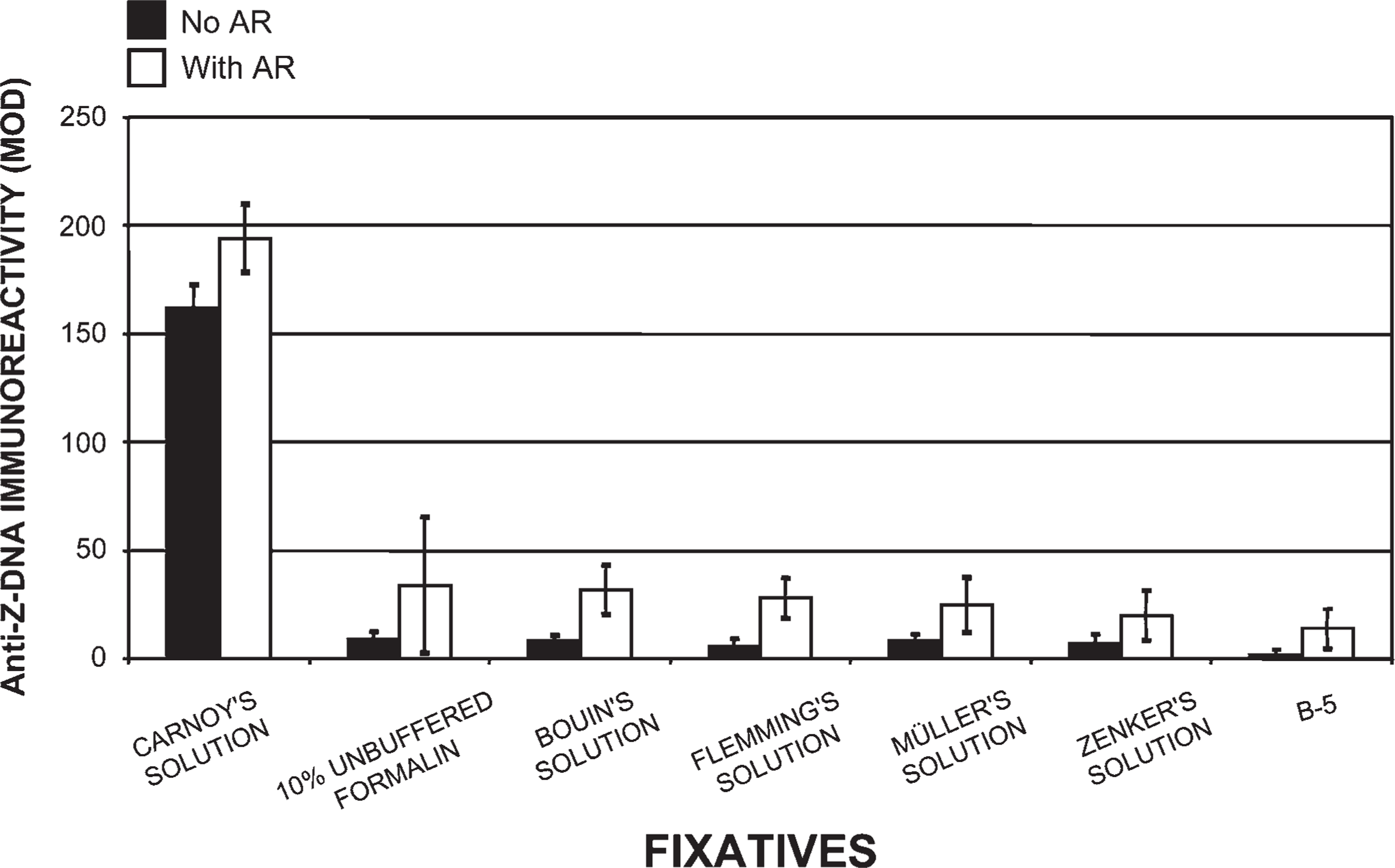
Effects of fixatives on Z-DNA immunoreactivity. Anti-Z-DNA (Z-4255 PAb) immunoreactivity (DAB) was quantified as MOD units. Our novel histotechnological processing procedure was used. All fixatives were compared with Carnoy's solution. Fixatives were used to characterize their effects on the content and structure of tissue-bound Z-DNA (1.5-μm-thick sections). This figure shows Z-DNA immunoreactivity (DAB: MOD) before and after 45% acetic acid treatments (i.e., AR). Seven different fixatives were used: Carnoy's solution, 10% unbuffered formalin, Bouin's solution, Flemming's strong solution, Müller's solution, Zenker's solution, and B-5 fixative. Forty five percent acetic acid pretreatments prior to antibody staining significantly increased MOD binding values in tissue fixed in Carnoy's solution, but it only slightly enhanced the manifestation of Z-DNA in the other six fixatives.
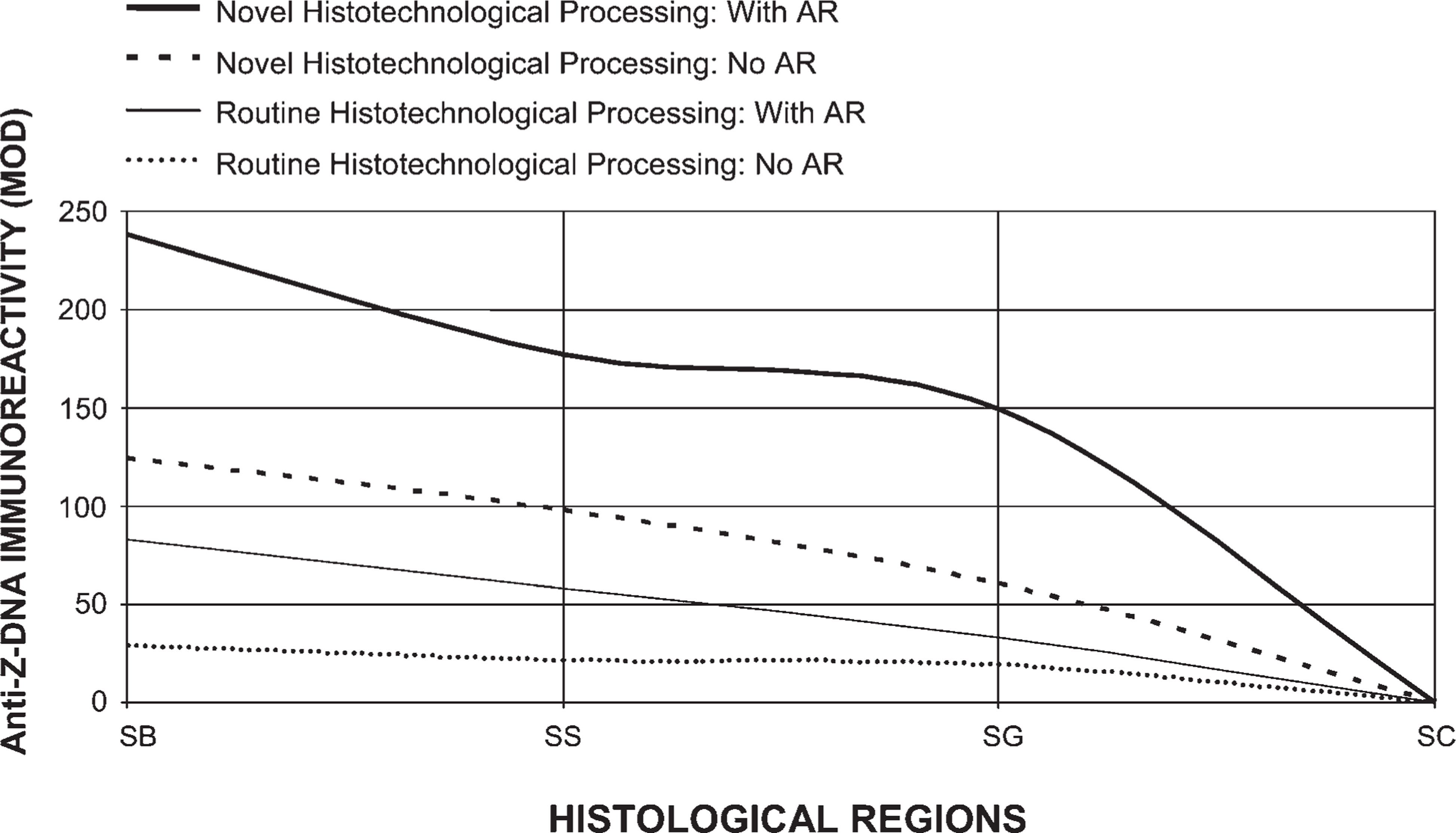
Comparison of Z-DNA immunoreactivity (DAB) with tissue (1.5-μm thick) processed by conventional procedures vs our histotechnological processing approach (with and without 45% acetic acid pretreatments: i.e., AR). Tissues were fixed in Carnoy's solution for optimal preservation of ds-DNA. This figure reveals how the quantification of Z-DNA MOD binding values can be improved by using our novel histotechnological processing procedure. Z-DNA immunoreactivity (DAB: MOD) with tissue processed using our histotechnological procedure (with 45% acetic acid pretreatments) compared with conventional processing (without 45% acetic acid pretreatments) resulted in a 717% increase in MOD binding values. The epidermis is divided into four parts, viz., SB, SS, SG, and SC.
Discussion
Our research indicates that using PicoGreen as a probe to stain fixed tissue sections is a more sensitive, cost-effective, and practical method for quantifying tissue-bound ds-DNA compared with other histologcal (e.g., aceto-carmine, aceto-orcein), histochemical (e.g., Hoechst 33258, acridine orange), and histotechnological techniques (e.g., Feulgen reaction), as well as immunohistochemical (e.g., anti-DNA antibody) DNA probes.
Use of PicoGreen, along with both of our new procedures, produces significantly more accurate qualitative and quantitative data, which allows for superior characterization of DNA (Figure 1–Figure 7). Our histotechnological processing approach and PicoGreen staining technique necessitate the use of ultrapure chemicals, fixatives, and water, as well as fast acquisition of tissue samples (i.e., reduced prefixation time), along with enzymatic AR and an ultraclean processing environment to preserve intact, undamaged, tissue-bound ds-DNA from degradation (viz., ss-DNA formation). Use of a microwave–vacuum oven enhances chemical reactions, speeds tissue penetration, lowers heating temperatures, and reduces histological processing times, which better stabilizes proteins and DNA in the fixed tissue samples. The choice of fixative and fixation time is absolutely critical for proper processing of undamaged, intact, tissue-bound ds-DNA. The combination of our histotechnological processing approach and PicoGreen staining procedure allows for maximum retention of unaltered ds-DNA content and DNA structure. This is evidenced by comparing tissue processed by conventional methods to our novel tissue-processing approach (Figure 7 and Figure 11). Use of Carnoy's solution, a microwave–vacuum oven, and reduced heating and light result in optimal PicoGreen staining.
Delaying fixation of postmortem tissue by 24 hr results in an increase of autolysis and anoxia, resulting in lower PicoGreen tissue-staining values. The quantity and quality of intact ds-B-DNA in fixed tissue sections was significantly affected by the time interval between tissue removal and placement into a fixative, especially for Z-DNA. Light negatively affects the PicoGreen probe; therefore, staining should be performed in the dark (or very dim light). Z-DNA is also very sensitive to light, and tissue sections on glass slides should not be exposed to excessive light prior to PicoGreen staining. Major biochemical changes occur in tissues within 10 min after anoxia (Kingsbury et al. 1995). Mitotic figures are reduced by 30%–50% because of a delay in tissue fixation of 2–6 hr (Cross et al. 1990). Prolonged tissue hypoxia reduces pH in tissues, which can directly decrease the amount of ds-DNA (Kingsbury et al. 1995). Approximately 30% of DNA and RNA can be lost during fixation (Srinivasan et al. 2002). Improper tissue fixation can lead to the degradation of ds-DNA due to nicking, which produces fragmented, denatured ss-DNA (Gagna et al. 1997). Preliminary data using anti-ss-DNA PAb probes to stain epidermal tissue sections shows that delayed fixation time of 24 hr causes an increase in denatured ss-DNA (∼41%) (data not shown). Damage to nucleic acids can interfere with the correct molecular characterization of fixed tissue-bound ds-DNA, especially involving cell death studies such as apoptosis. Therefore, biopsy or surgically removed animal tissue must be diced quickly into small pieces and placed immediately into a fixative. Another consideration is that the anesthetic and euthanizing agents may interfere with DNA probe staining. Other preliminary data have shown that our histotechnological processing method allowed for extraction of superior quality DNA (i.e., higher molecular weight) from fixed, paraffin-embedded tissue blocks (data not shown). Our reduced prefixation time (2 min or less) is critical in preserving DNA, especially for less stable alternative types of nucleic acids such as Z-DNA.
For DNA studies, use of organic solvents or alcohol fixatives is superior to aldehyde fixatives (Hopwood 1970; Bramwell and Burns 1988; Hobot and Newman 1991; Douglas and Rogers 1998; Serth et al. 2000). If properly used, precipitating fixatives such as Carnoy's solution, methacarn, and Clarke's solution will yield superior fixation and retention of undamaged tissue-bound ds-DNA (Figure 1–Figure 11). Therefore, use of these fixatives along with microwave–vacuum oven technology and our histotechnological processing procedure will produce superior tissue sections for staining with PicoGreen. The proper choice and specific use of a fixative can prevent or significantly reduce the use of AR. Use of enzymatic AR prior to fixation is not suggested because it leaves tissue susceptible to disintegration and the loss of ds-B-DNA, especially Z-DNA. From our data, Carnoy's solution, methacarn, and Clarke's solution are the preferred fixatives for tissue-bound DNA studies. The majority of DNA fixation using these three solutions occurs via entrapment of the ds-DNA by the fixation of surrounding nuclear proteins. These fixatives contain acetic acid, which is a rapid non-coagulant that precipitates nucleoproteins differently than non-nuclear proteins. Alcohols are cytoplasmic coagulants, but they do not fix chromatin. If fixation is prolonged, non-aqueous chemicals such as alcohols can extract DNA from processed tissue (Bramwell and Burns 1988; Douglas and Rogers 1998). Methanol and ethanol can change the structure of proteins by disrupting hydrophobic interactions that maintain tertiary structures. These alcohols may preserve the secondary structure of proteins and associated epitopes by stabilizing their hydrogen bonds. The reason why Carnoy's solution allows for higher PicoGreen staining than Clarke's solution (Figure 3) is due to the addition of chloroform. Chloroform speeds the process of fixative penetration, denatures DNA binding proteins, and exposes more ds-DNA. The potential for chloroform binding to DNA is very low (Rosenthal 1987).
The use of a microwave–vacuum oven at 35C to 40C for fixation and histotechnological processing of tissues produces superior preservation of ds-DNA content and structure by increasing the speed of fixation, reducing the exposure of tissue and DNA to heat and reducing enzyme degradation of ds-DNA (Marani et al. 1996). Simultaneously microwaving and vacuuming accelerates chemical fixation by increasing the penetration and chemical reactions of the fixative, especially when using a fast-penetrating fixative such as Carnoy's solution. Microwave–vacuum oven fixation is considered both chemical and thermal coagulation. Low-power microwave energy together with the ColdSpot eliminate specimen heating (Giberson and Demaree 2001). Low microwave wattage (e.g., 150 W) favors diffusion in tissue, whereas high wattage (e.g., 650 W) favors shifting the chemical equilibrium that normally exists in tissue (Giberson and Demaree 2001). Many histological protocols involve heating tissues to 80C or 100C (Giberson and Demaree 2001; Henwood 2005). However, heating tissue to 50C destroys proteins (Srinivasan et al. 2002). Mammalian genome DNA denatures with a Tm of ∼87C–90C (Marmur and Doty 1961), with changes in DNA structure occurring at 73C–86C (data not shown).
Throughout this paper we have emphasized the use of ultrapure chemicals and water, as well as clean working areas for all procedures. Dimmed lights are also critical during the PicoGreen tissue-staining process. Unspecific compounds, contaminants, overfixation, DNA denaturation, and cross-linking elements negatively affect PicoGreen binding. We believe that even the slightest impurities will negatively affect the quantitative ds-DNA data. Normal, technical-grade formaldehyde may contain formic acid and additives that will interfere with the ability of PicoGreen to bind to ds-DNA. Many forms of glutaraldehyde and formaldehyde contain cyclical polymers and impurities (Gillett and Gull 1972), which may also denature the ds-DNA. Use of highly purified reagents (e.g., EM-grade fixatives) that are free of contaminants allow for superior preservation and quantification of tissue-bound ds-DNA (Hobot and Newman 1991). Additionally, residual paraffin blocks DNA binding sites and may not be completely removed by AR.
We believe that paraffin-embedded tissues that are immediately fixed in 10% unbuffered formalin, then sectioned and stored at room temperature for 8 years, resulted in lower PicoGreen binding primarily due to ds-DNA that was overfixed (i.e., 72 hr). A decrease in PicoGreen was mainly due to excessive cross-linking that was irreversible, even with AR. During the storage period, ds-DNA may have also been degraded by oxidation. All of these factors caused problems with preserving ds-DNA content and structure. Our other two experiments (viz., delay of 24 hr in fixation and conventional processing vs our novel histotechnological procedure) provide further proof that tissue must be quickly fixed (i.e., prefixation time of 2 min or less) and properly processed to obtain optimal characterization using PicoGreen for quantifying ds-DNA and, especially, Z-DNA. We believe that much of the scientific community's archival tissue blocks are not suited for many molecular studies.
Of all the cross-linking solutions, the best aldehyde fixative is zinc formalin because it produced results closest to the alcohol fixatives, did not require extensive AR, and yet still produced superior tissue morphology (Figure 4). Zinc sulfate helps to preserve tissue antigenicity and usually eliminates or reduces the need for AR. Zinc ions stabilize the tissue proteins, preventing conformational changes and cross-linking caused by formaldehyde. Except for the zinc formalin, Davidson's and FAA fixatives required the least amount of AR of any other fixative containing formalin (Figure 3 and Figure 4) because they contain precipitating chemicals. The combination of cross-linking and precipitating chemicals results in an excellent fixative for the retention of DNA content and structure, requiring less AR. STF allows for superior quantitative (Figure 3) and, especially, qualitative results, with only minimal AR. STF, which is a non-cross-linking fixative free of alcohol, formalin, RNase, and DNase, produces excellent tissue sections and eliminates or reduces the need for AR.
Fixatives that caused the most problems for PicoGreen staining, requiring extensive AR and producing the lowest MOD binding values, were Bouin's solution, Zenker's solution, Flemming's strong solution, Müller's fluid, and B-5 fixative (Figure 6), as well as 1% glutaraldehyde, 10% ultrapure NBF, and 10% unbuffered formalin (Figure 4 and Figure 5). Bouin's solution has an acidic environment that damages both DNA and RNA. The first five fixatives contain mercuric chloride, potassium dichromate, picric acid, chromic acid, and osmium tetroxide, which probably block binding sites on the ds-DNA, thereby preventing PicoGreen from interacting with the ds-DNA and/or directly degrading the ds-DNA into denatured low molecular weight ss-DNA that is nicked and fragmented. Heavy metals found in some of these fixatives are compromising the structure of ds-DNA. B-5 fixative and Bouin's solution have been shown to be very poor fixatives for DNA preservation (Srinivasan et al. 2002). Even when tissues are washed to remove these metals, they still interfered with PicoGreen staining (Figure 6). This type of steric interference can directly block binding sites on tissue-bound ds-DNA (such as major and/or minor grooves, bases, and the phosphodiester backbone) or interact with DNA-protein complexes.
Cross-linking fixatives are among the most popular fixatives used by scientists. Some laboratories store tissue in solution, such as 10% NBF or formalin, for long periods of time. This may be good for routine histological processing, but it is not acceptable for molecular characterization of tissue-bound ds-DNA. Fixation, especially overfixation, with commonly used aldehyde fixatives (i.e., the cross-linking fixatives used in this study) produces extensive cross-links between DNA and proteins and between proteins and proteins and may also form protein–DNA–formaldehyde complexes (Hopwood 1970; Gagna et al. 1997; Serth et al. 2000). The formaldehyde itself may be blocking the DNA binding sites from the PicoGreen. Tissue fixation in high concentrations of glutaraldehyde (>1%)for 15 min or more is detrimental to antigenic response (Hobot and Newman 1991). Another problem is prolonged exposure of tissue to formic acid or non-neutral formalin (e.g., 10% unbuffered formalin with a pH of 3.0 or greater) (Figure 5) because it causes degradation of ds-DNA. During prolonged exposure, intact high molecular weight ds-DNA is converted to damaged denatured low molecular weight ss-DNA fragments, which alters DNA content, structure (e.g., Z-DNA), and cellular location, and produces incorrect data (Gagna et al 1997). Characterization of Z-DNA requires processing in alcohol fixatives. “Masking” of B-DNA and/or Z-DNA binding sites by non-alcohol fixatives (e.g., 10% NBF) produces cross-links that may not be fully removed even with enzymatic digestion or 45% acetic acid.
Chemical reactions between formaldehyde and DNA have been characterized by many investigators (Hopwood 1970; Bramwell and Burns 1988; Hobot and Newman 1991; Douglas and Rogers 1998; Serth et al. 2000). Formaldehyde-based fixatives can cause significant denaturation of tissue-bound ds-DNA, such as formation of ss-DNA, by causing interchain hydrogen bond breaks and base unstacking. Formaldehyde also causes slow hydrolysis of the phosphodiester bonds of ds-DNA (Srinivasan et al. 2002). The rate at which modifications of ds-DNA by formaldehyde take place is dependent on temperature, concentration, impurities, time of exposure, and pH. Formaldehyde fixation causes formaldehyde-induced histone-DNA cross-links. Short fixation treatment produces reversible covalent bonds between DNA and histones (Brutlag et al. 1969), which may be removed by AR (Roth et al. 2000; Shi et al. 2000). Unlike acetic acid that penetrates rapidly, formalin has a moderate speed of tissue penetration so its fixation action is slow. Regarding DNA, formaldehyde fixation occurs by mechanical retention due to fixation of the surrounding material. In this situation, AR must be used to expose all DNA binding sites. Formaldehyde, the smallest of the aldehydes, penetrates tissue faster than glutaraldehyde, which is a much larger molecule. Formaldehyde provides a mild fixation, whereas glutaraldehyde provides more rigid cross-linking of proteins and better entrapment of DNA (Hopwood 1970; Bramwell and Burns 1988; Hobot and Newman 1991). The negative consequences of cross-linking include steric hindrance of the diffusion of large molecules such as embedding media (e.g., paraffin wax) and PicoGreen staining.
Use of anti-Z-DNA PAb and MAb reveals that our novel histotechnological processing procedure greatly increased retention of total DNA content and aided in retaining specific DNA structures and/or the potential for specific DNA conformations (i.e., helical transitions) such as right-handed ds-B-DNA to left-handed ds-Z-DNA. The ratio of B-DNA to Z-DNA in fixed tissue sections is usually ∼7 to 8 (Gagna et al. 1997). Our results concur with these numbers (Figure 9 and Figure 10). By retaining more undamaged, intact, right-handed ds-B-DNA, we allow for a greater potential of B-DNA to Z-DNA transitions (Gagna et al. 1997; Herbert and Rich 1999; Rich and Zhang 2003). We speculate that the mechanism behind an increase in Z-DNA immunoreactivity following 45% acetic acid pretreatment (AR procedure), prior to antibody staining, is due to the low pH. Use of 45% acetic acid removes Z-DNA binding proteins, exposes existing Z-DNA, and/or removes B-DNA binding proteins, allowing for B-DNA to Z-DNA transitions. Forty five percent acetic acid removes core histones from deoxyribonucleoprotein, thereby releasing negative superhelical turns into freed ds-DNA. The energy of deformation associated with negative supercoiling allows for the conversion of B-DNA into Z-DNA (Gagna et al. 1997; Herbert and Rich 1999; Rich and Zhang 2003). Other mechanisms by which 45% acetic acid may cause the B-DNA to Z-DNA transition is by protonation of the bases and/or phosphates and by strand separation (Gagna et al. 1997). Acetic acid, a component of Carnoy's solution, removes chromosomal proteins (e.g., histones) and non-chromosomal proteins better than ethanol (Gagna et al. 1997). Extraction of histones, especially histone H1, allows for the optimal staining of ds-Z-DNA (Gagna et al. 1997). Production of denatured ss-DNA prevents ds-DNA negative supercoiling from initiating a B-DNA to Z-DNA transition (Gagna et al. 1997; Herbert and Rich 1999; Rich and Zhang 2003). Formaldehyde fixatives may prevent the full removal of core histones and/or damage ds-DNA, limiting Z-DNA exposure and/or formation. Therefore, any damage to ds-DNA during histological processing will negatively affect the quantification of left-handed ds-Z-DNA.
We believe that the PicoGreen probe binds to tissue-bound, right-handed ds-B-DNA, not left-handed ds-Z-DNA. No previous evidence concerning this issue exists. Preliminary data of solution studies reveal that PicoGreen binds strongly to B-DNA, but not to Z-DNA (data not shown). The precise mechanism by which PicoGreen binds to ds-DNA is not fully understood. It is speculated that it interacts via base-selective groove binding and/or by charges, not by classic intercalation. PicoGreen is positively charged, which may cause interactions with the negative charge of the phosphodiester backbone of DNA. Additional proof that this probe is binding to B-DNA and not to Z-DNA can be found in signal studies that indicate PicoGreen prefers A–T over C–G base pairs (Haugland 2002). Z-DNA sequences develop predominantly with C–G base pairs (Gagna et al. 1997).
Our results prove that alcohol fixatives (e.g., Carnoy's solution, methacarn, and Clarke's solution) are superior to aldehyde fixatives (e.g., zinc formalin, 1% glutaraldehyde, 10% ultrapure NBF, and 10% unbuffered formalin), aldehyde–alcohol fixatives (e.g., FAA and Davidson's), and commercially available fixatives (e.g., STF) when used with our novel histotechnological procedure for preserving both B-DNA and Z-DNA (Figure 1–Figure 11). All other fixatives (viz., Bouin's, Zenker's, Flemming's, Müller's, and B-5 solutions) should be avoided when quantifying ds-B-DNA and Z-DNA (Figure 6 and Figure 10). Basically, the same results that occurred for B-DNA occurred for Z-DNA (Figure 7–Figure 11). It should be noted that a slight overfixation in aldehyde fixatives can possibly be remedied by AR. However, overfixation in alcohol fixatives, which causes a loss of DNA, cannot be corrected by AR. The investigator may have to modify certain procedures based on the type of tissue. If used with both of our approaches, Carnoy's solution retains more intact unaltered ds-DNA and requires less AR than other fixatives. Therefore, it allows for the highest PicoGreen binding. Carnoy's solution also yields excellent tissue morphology when using our histotechnological processing procedure. Additionally, alcohol fixatives allow for superior exposure of alternative forms of nucleic acids such as ds-Z-DNA.
Fixed tissues provide a very important source of DNA for molecular biologists and pathologists. With the growth of histochemistry, immunohistochemistry, in situ hybridization, molecular histotechnology, laser-capture microdissection, tissue culture, tissue micro-arrays, and DNA extraction from archival tissue blocks, knowledge about the effects of fixation and tissue processing on both the utility and integrity of the preserved nucleic acids and nucleic acid–protein complexes is extremely important (Kiernan and Mason 1996; Carson 1997; Bowtell and Sambrook 2003). Fluorescent microspectroscopy of tissue sections using our novel PicoGreen ds-DNA staining technique and histotechnological processing approach far outweighs any inconveniences and will create new research possibilities for scientists who characterize the structure and function of fixed tissue-bound ds-DNA. Our data show that conventional histological processing results in damaged and/or altered DNA tissue-bound content, as compared with tissue processed by our novel technique. These new approaches will allow for increased retention of DNA content, DNA location, DNA structure (e.g., B-DNA, Z-DNA), superior qualitative analysis and, especially, quantification of tissue-bound, intact, unaltered high molecular weight ds-DNA. These approaches will also allow for superior data obtained from downregulation of genes, the aging process, alternative DNA structures, apoptosis, terminal differentiation, denucleation, necrosis, and DNA ploidy analysis. Having characterized 15 fixatives will help researchers make better choices for tissue processing. Our novel fluorescent DNA staining protocol is a rapid, reproducible, and cost-effective method that will enhance molecular histochemistry in biomedical research, drug discovery and development, forensic analysis, and diagnostic molecular pathology.
Footnotes
Acknowledgements
The research was supported in part by an American Association of University Professors (AAUP)/New York Institute of Technology (NYIT) research grant and an NYIT Institutional Support for Research and Creativity Grant.
The authors thank Susan Law, Amy Law, Tina Law, Neha N. Khariwala, Asima N. Ahmad, Anetta Raysin, and Noor ul ain Hashmi for their help in preparing this manuscript.