
Abstract
Detection of specific nuclear transcripts by fluorescence in situ hybridization (FISH) has constituted a major breakthrough in the study of the organization of transcription in the cell nucleus. Using the model of heat shock genes, we present an optimized procedure for nuclear transcripts that provides high efficiency for RNA detection and good preservation of cell morphology and nuclear texture. Using this procedure, we designed an original high-efficiency methodology combining FISH and fluorescence immunocytochemistry (FICC), which is used here for the simultaneous detection of heat-shock protein (hsp) nuclear transcripts and the specific heat-shock transcription factor 1 (HSF1). We show that the nuclear accumulation sites of HSF1 in heat-shocked cells do not correspond to the sites of transcription of the hsp70 gene.
Keywords
T
We were interested in combining FISH and FICC to simultaneously detect nuclear transcripts and specific transcription factors. Several procedures with substantial different efficiency for the detection of nuclear transcripts have been reported thus far (Lawrence et al. 1989; Zirbel et al. 1993; Xing et al. 1995; Clemson et al. 1996; Dirks et al. 1997). On the basis of the comparison of four existing procedures, we have developed an original optimized procedure. Three criteria were taken into consideration to evaluate the procedures: (a) preservation of cell morphology, (b) preservation of nuclear texture, and (c) efficiency for RNA detection. Using this optimized protocol for nuclear transcript detection, we designed a rapid and high-efficiency procedure combining FISH and FICC to simultaneously detect specific nuclear transcripts and transcription factors.
This study was performed using the model of heat-shock genes (hsp) (Schlesinger 1990). hsp genes are abundantly transcribed on heat shock (Morimoto 1994), thus facilitating the detection of nuclear transcripts by FISH. Moreover, the heat-induced expression of hsp genes is controlled by the specific heat-shock transcription factor 1 (HSF1), which is concentrated in large foci in the nuclei of heat-shocked cells (Sarge et al. 1993). This model is therefore well suited to study the molecular events that occur during gene activation. The procedure we present here was applied to study the relative distribution of HSF1 and hsp70 gene transcription sites.
Materials and Methods
Cell Culture and Heat Shock
Normal primary fibroblasts (skin) were grown directly on glass slides in RPMI medium (Gibco BRL; Gaithersburg, MD) supplemented with 10% fetal calf serum. Heat shock was performed in a water bath for 1 hr at 44C.
Probes and Antibodies
hsp70 transcripts were detected using the genomic probe pH2.3 (a gift from Dr. R.I. Morimoto, Evanston, IL), covering the entire coding sequence of the intronless hsp70 gene (2.3
Fluorescence In Situ Hybridization
Four procedures differing in the type of permeabilization treatments used to improve accessibility to the target were tested for the detection of nuclear transcripts. The simplest procedure uses no permeabilization (Protocol 1). Two other procedures use different postfixation permeabilization steps (Protocols 2 and 3), and the fourth one uses pre- and post-fixation permeabilizations (Protocol 4). All these procedures are summarized in Table 1. Unless otherwise noted, all incubations were performed for 5 min.
Protocol 1
This protocol is derived from Lawrence et al. (1989).
Cell Treatment. Immediately after heat shock, cells were fixed in 4% formaldehyde/PBS (pH 7) for 10 min and dehydrated through ethanol baths. If 3D analyses have to be performed, dehydrations can be omitted and replaced by an equilibration step in 50% deionized formamide/2 x SSC.
Hybridization. A total of 100 ng of labeled probe was precipitated with 10 μg of salmon sperm DNA. The DNA pellet was resuspended in 10 μl of 50% deionized formamide/10% dextran sulfate/2 x SSC, denatured for 5 min at 75C, and applied to the dry slide. Hybridization was allowed to proceed overnight at 37C. Cells were then washed in 60% formamide/2 x SSC, once at 45C and twice at room temperature (RT), and rinsed three times in 2 x SSC at RT. After a 30-min incubation at 37C in a blocking buffer (3% BSA/0.3% Triton X-100/4 x SSC), the probe was detected using either avidin-FITC (Vector; Burlingame, CA) or an anti-digoxigenin antibody coupled to TRITC (Boehringer; Mannheim, Germany). Cells were then washed three times in 4 x SSC/0.1% Tween-20 at 42C and embedded in an anti-fading solution consisting of 90% glycerol, 20 mM Tris-Cl, 2.3% 1,4-diazobicyclo-octane (DABCO; Sigma, St Louis, MO), 0.02% NaN3, and DAPI (250 ng/ml) as a DNA counterstain.
Protocol 2
This procedure derives from Zirbel et al. (1993). After formaldehyde fixation, accessibility of the probe to the target was improved by a 10-min incubation in 0.1 M HCl, followed by two successive incubations of 10 min each in 0.5% saponin/0.5% Triton X-100/PBS. As previously reported (Clemson et al. 1996), prolonged incubations in the detergent mixture ensure maximal nuclear RNA detection. Dehydration and hybridization are the same as in Protocol 1.
Fixation and permeabilization steps for each protocol that was tested for RNA detection a
aUnless otherwise stated, all incubations are performed at room temperature. Hybridization and detection steps are performed as described in Materials and Methods.
bSee composition in Materials and Methods.
Protocol 3
This protocol combines Protocol 2 with the procedure from Popp et al. (1990) using liquid nitrogen. After HCl and saponin/Triton permeabilizations, cells were equilibrated in 20% glycerol/PBS for 20 min and freeze-thawed three times by briefly dipping in liquid nitrogen. Cells were then rinsed in PBS, dehydrated, and hybridized as described in Protocol 1.
Protocol 4
This protocol combines Protocol 2 with a procedure derived from van den Engh et al. (1984), using an additional prefixation permeabilization step. Immediately after heat shock, cells were incubated for 10 min at RT in a nucleus isolation buffer (3 mM dithiothreitol, 5 mM Hepes, 50 mM KCl, 10 mM MgSO4) and subsequently incubated for 10 min at −20C in the same buffer supplemented with 0.25% Triton X-100. Cells were then rinsed briefly in PBS and fixed in 4% formaldehyde/PBS for 10 min. Other steps are identical to Protocol 2.
Fluorescence Immunocytochemistry
HSF1 was detected according to the procedure described by Sarge et al. (1993). Cells were fixed for 10 min in 4% formaldehyde/PBS, incubated for 45 min at 37C in a blocking buffer (10% FCS/0.3% Triton X-100/PBS), and subsequently incubated for 90 min at 37C with the rat monoclonal anti-HSF1 antibody. After three washes at 42C in 2% FCS/0.3% Triton X-100/PBS, detection was performed using a sheep anti-rat antibody coupled to FITC (Sigma) and nuclei were counterstained with DAPI.
Combined FICC and FISH
The best results were obtained when FISH was performed before FICC (see Results and Discussion). Specific nuclear transcripts were detected according to Protocol 3 as described above. Detection of transcript-specific probes labeled with biotin was performed using avidin-FITC. After post-detection washes in 4 x SSC/0.1% Tween-20, a 45-min incubation in 10% FCS/0.3% Triton X-100/PBS was performed, followed by incubation for 90 min at 37C with the rat monoclonal anti-HSF1 antibody. Detection was performed using a sheep anti-rat antibody coupled to TRITC (Sigma), and nuclei were counterstained with DAPI.
Simultaneous Detection of DNA and RNA Sequences
hsp70 transcripts were detected with the pH 2.3 probe, together with a cosmid probe specific for the 6p21.3 locus (Burfeind et al. 1994). The simultaneous detection of DNA and RNA sequences was performed in a single-step FISH experiment as previously described (Clemson et al. 1996). The only difference with nuclear transcript detection resides in the denaturation step. Briefly, 100 ng of the cosmid probe and 300 ng of the cDNA probe were precipitated together with 3 μg of human Cot I competitor DNA, denatured at 75C for 5 min, and incubated for 1 hr at 37C to allow suppression of repeated sequences. Cells were permeabilized after fixation according to Protocol 3 and subsequently denatured by a 1-min incubation in 70% formamide/2 x SSC at 75C, followed by a 30 sec incubation in 50% formamide/2 x SSC at the same temperature. The denatured probes were applied to the slide. Hybridization and detection steps were performed as described above for nuclear transcript detection.
Digital Imaging
Preparations were observed under an epifluorescence microscope (Zeiss Axiophot; Oberkochen, Germany) equipped with a 100-W mercury lamp using appropriate filter sets. Images were acquired with a cooled CCD camera (C4880 Hamamatsu; Tokyo, Japan) using a x63, 1.25 NA oil immersion objective with an intermediate magnification of x1.25. All images were acquired with the same exposure time to allow comparison of hybridization signals.
Results and Discussion
Optimization of Nuclear Transcript Detection by FISH
We present here an optimized procedure for nuclear transcript detection that was developed on the basis of a comparison of four procedures described in the literature (van den Engh 1984; Lawrence et al. 1989; Popp et al. 1990; Zirbel et al. 1993). These procedures differ in the nature of the permeabilization steps used to improve the accessibility of the probe to its nuclear target (summarized in Table 1). Protocol 1 does not use any permeabilization step before hybridization, Protocols 2 and 3 include permeabilizations after fixation, and Protocol 4 uses permeabilizations before fixation. To our knowledge, only Protocols 1 and 2 have been used up to now for the detection of nuclear transcripts. Hsp90α and hsp70 nuclear transcripts were detected using these procedures in non-heat-shocked and heat-shocked primary fibroblasts, and the protocols were compared on the basis of the following criteria: (a) efficiency for RNA detection, (b) preservation of cytoplasmic RNAs, and (c) preservation of nuclear texture.
Efficiency of the Different Procedures for RNA Detection
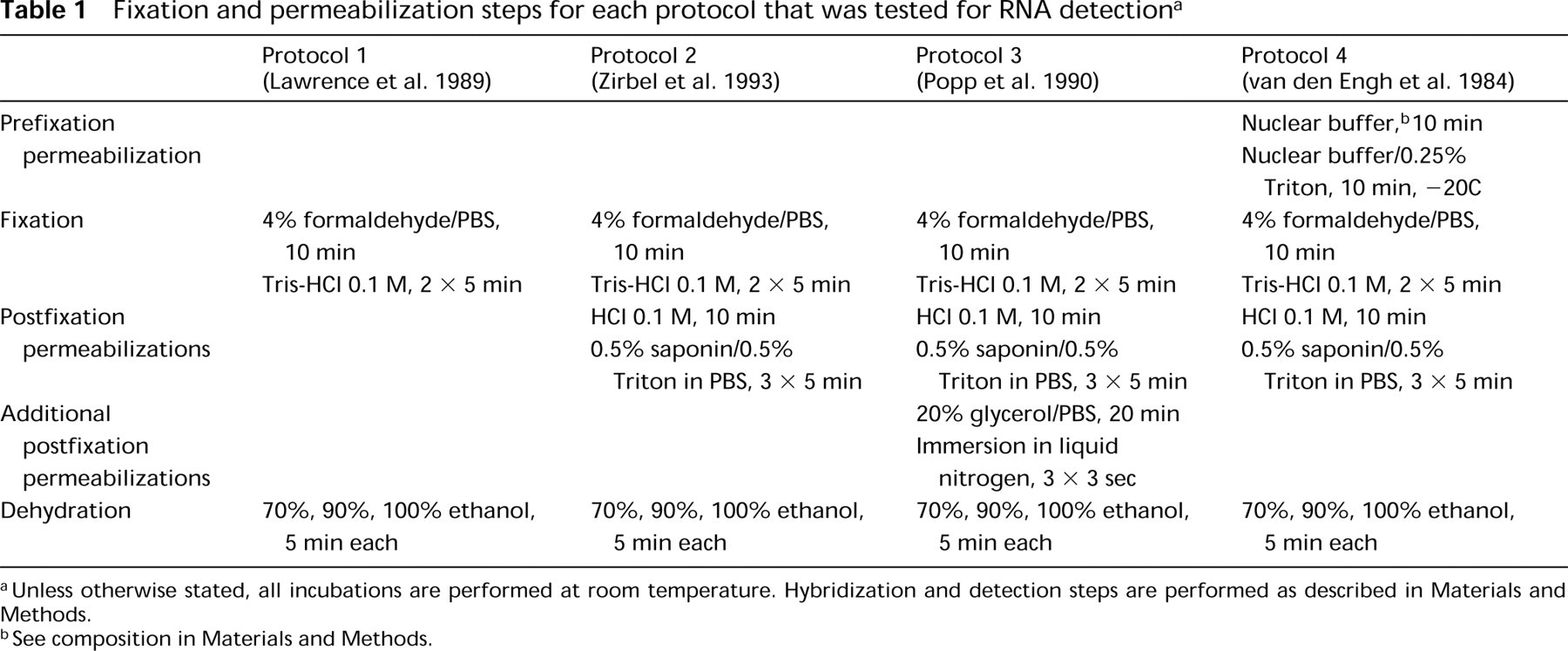
In the absence of permeabilization treatment (Protocol 1), two foci corresponding to nuclear accumulations of hsp90α (Figure 1A) and hsp70 transcripts (Figure 1B) were detected in all heat-shocked cells. Although the hsp90α gene, in contrast to the hsp70 gene, is known to be constitutively expressed in these cells, no hybridization signals could be detected in unstressed cells (data not shown). As previously demonstrated by others for other transcripts, nuclear transcript foci represent the sites of transcription of the corresponding genes (Moen et al. 1995). In a previous study, we have shown that hsp70 nuclear transcripts were always found in the vicinity of chromosome 6 centromeres (Jolly et al. 1997), as expected from the localization of the hsp70 gene in the 6p21.3 region (Harrison et al. 1987). In addition, co-detection of hsp70 (Figure 1P) or hsp90α (data not shown) transcripts with a cosmid probe specific for the gene locus revealed a close spatial proximity between nuclear transcripts and their corresponding gene loci.
When postfixation permeabilization steps with HCl and saponin/Triton were added (Protocol 2), nuclear foci corresponding to hsp90α (Figure 1D) and hsp70 transcripts (Figure 1E) were significantly larger and brighter than the signals obtained with Protocol 1 (compare with Figures 1A and 1B). When an additional postfixation permeabilization step with liquid nitrogen was used (Protocol 3), hybridization signals corresponding to both hsp transcripts appeared larger than with Protocol 2 (compare Figures 3G and 3H with Figures 3D and 3E). In addition, hsp90α transcripts could be detected in non-heat-shocked cells using either Protocol 2 (data not shown) or Protocol 3 (Figure 1M). As expected from the lower constitutive expression of the hsp90α gene (Hickey et al. 1986), the nuclear foci were much smaller in unstressed cells than in heat-shocked cells (compare Figures 1M and 1G). No foci could be detected for the hsp70 gene, whose constitutive expression is extremely low (Figure 1N) (Wu et al. 1985).
When a prefixation permeabilization step in a Triton-containing buffer was added to the HCl and saponin/Triton permeabilizations (Protocol 4), hybridization signals corresponding to hsp90α (Figure 1J) and hsp70 (Figure 1K) transcripts were found to be identical in size to those obtained with Protocol 3 (compare with Figures 1G and 1H). The use of Triton before fixation has been shown to significantly improve the efficiency of nuclear RNA detection (Clemson et al. 1996; Dirks et al. 1997). As with Protocol 3, hsp90α transcripts could also be detected in non-heat-shocked cells (data not shown).
These observations clearly show that pre- and post-fixation permeabilization steps greatly improve the efficiency of nuclear RNA detection.
Preservation of Cytoplasmic RNAs
In addition to the focal concentrations of hsp transcripts, diffuse labeling was also detected in the nucleoplasm of non-heat-shocked and heat-shocked cells with all procedures (Figures 1A–1K). Diffuse staining of lesser intensity was also observed in the cytoplasm, but only with Protocols 1, 2, and 3 (Figures 1A and 1H). In contrast, no cytoplasmic labeling was detected using Protocol 4 (compare Figures 1J and 1K with Figures 1G and 1H). As previously suggested (Clemson et al. 1996), this absence of cytoplasmic labeling is most probably due to the use of Triton before fixation. Analysis of the preparation with a differential interference contrast microscope revealed that the cytoplasm of the cells was still present. The diffuse cytoplasmic and nucleoplasmic stainings are likely to represent diffusely distributed transcripts, as assessed by their absence when cells were treated with RNase A before hybridization (data not shown). These observations suggest that the cytoplasmic RNAs are not removed during postfixation permeabilizations but can be lost and/or digested during prolonged prefixation permeabilizations.
Preservation of the Nuclear Texture
The nuclear texture was investigated on the basis of DAPI counterstaining. With all four protocols, the overall nuclear texture was similar (Figures 1C, 1F, 1I, 1L, and 1O). In particular, the nucleoli and the Barr body were always clearly visible (arrows and arrowheads, respectively), suggesting that no major rearrangements in the nuclear organization were induced by pre- and postfixation permeabilizations.
Detection of hsp90a (left column) and hsp70 (middle column) nuclear transcripts in non-heat-shocked (
In conclusion, pre- and postfixation permeabilization steps significantly improve the efficiency of RNA detection, as assessed by measuring the size of the hybridization signals. Two procedures proved to be more efficient with respect to RNA detection, the one using liquid nitrogen after fixation (Protocol 3) and the one using a Triton extraction before fixation (Protocol 4). However, the procedure using liquid nitrogen as a postfixation permeabilization step appears to be the method of choice, because it combines maximal nuclear RNA detection and good preservation of the cellular and nuclear morphology. This procedure appears to be particularly interesting for the detection of poorly expressed RNAs and/or for investigation of the transcriptional activity of amplified genes in tumor cells, as previously described (Jolly et al. 1997).
Co-detection of Nuclear Transcripts and Transcription Factors
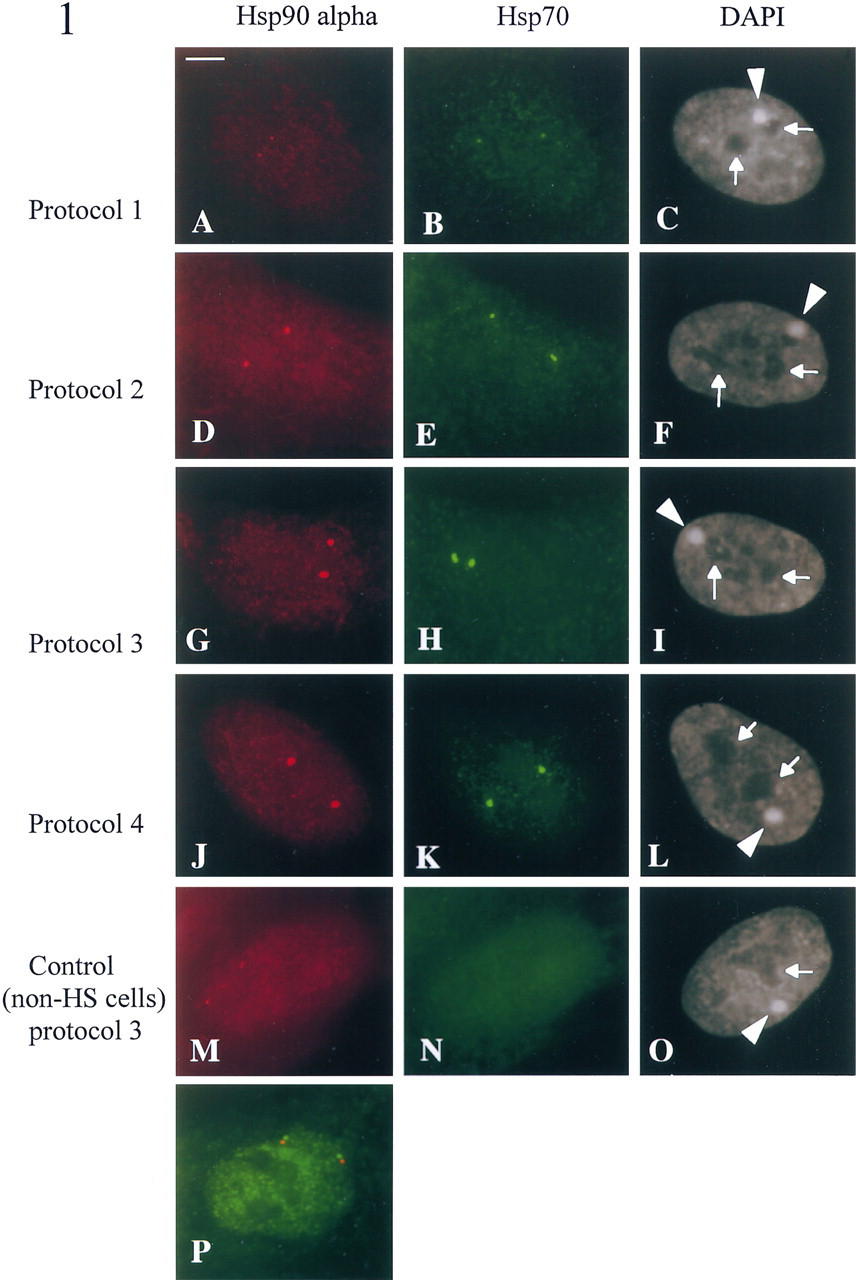
Detection of HSF1 by Fluorescence Immunocytochemistry. HSF1 was detected using a rat monoclonal anti-HSF1 antibody. As previously reported (Sarge et al. 1993), in non-heat-shocked cells HSF1 displayed a diffuse homogeneous staining, preferentially in the nucleoplasm and, to a lesser extent, in the cytoplasm (Figure 2A). In heat-shocked cells, HSF1 was found to concentrate in two large foci in each nucleus (Figure 2B) (submitted for publication). A diffuse staining, less intense than in control cells, persisted in the nucleoplasm; in contrast, no cytoplasmic labeling was detected in heat-shocked cells (compare Figures 2A and 2B). The absence of cytoplasmic labeling in heat-shocked cells is consistent with the heat-induced trans-location of HSF1 into the nucleus (Sarge et al. 1993).
The optimized procedure for nuclear transcript detection using postfixation permeabilization steps with HCl, saponin/Triton, and liquid nitrogen was chosen for combination with fluorescence immunocytochemistry. The impact of these treatments on the distribution of HSF1 factor in heat-shocked cells was thus investigated. As shown in Figure 2C, no significant difference in the size and intensity of nuclear HSF1 foci was observed compared to the original FICC procedure, which uses only Triton in the blocking and detection steps (Sarge et al. 1993). In addition, the diffuse nucleoplasmic labeling was also identical to that observed with the original procedure (compare Figures 2C and 2B). These observations show that the distribution of HSF1 is unaffected by postfixation permeabilization treatments.
Intracellular localization of HSF1 in non-heat-shocked (
Simultaneous detection of hsp70 nuclear transcripts (
Simultaneous Detection of HSF1 and hsp Nuclear Transcripts
Two different approaches were tested. Because FICC requires only mild permeabilization steps, it seemed a priori preferable to perform FICC first (FICC<FISH). Alternatively, because nuclear transcripts are highly sensitive to nucleases and may be digested during the FICC detection, a procedure in which FISH was performed first was also tested (FISH<FICC).
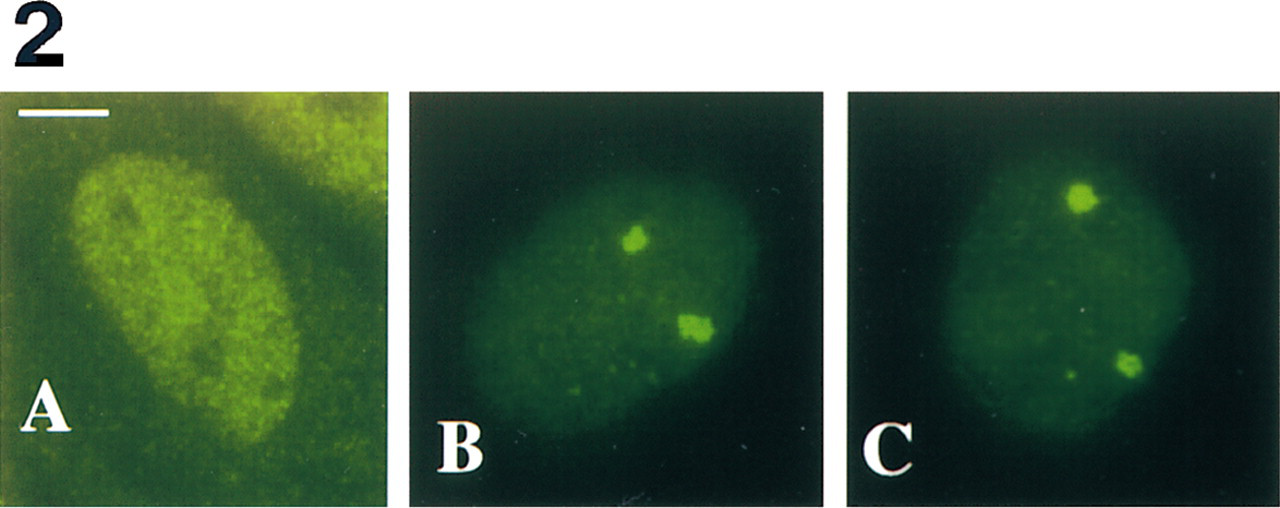
Illustrations of co-detection of HSF1 and hsp70 transcripts using either the FICC<FISH or the FISH< FICC procedure are shown in Figure 3. Signals corresponding to hsp70 nuclear transcripts (Figures 3A and 3C) and transcription factors (Figures 3B and 3D) were detected with both procedures. However, the intensity and size of the signals varied from one procedure to another. With the FISH<FICC procedure, fluorescent signals obtained for both transcripts and transcription factors were similar to those obtained when HSF1 or nuclear transcripts were detected independently (compare Figure 3A with Figure 1H and Figure 3B with Figure 2B). With the FICC<FISH procedure, the fluorescent signals corresponding to HSF1 foci (Figure 3D) appeared identical in shape and size to HSF1 foci observed with the FISH<FICC procedure (compare with Figure 3B). In contrast, the signals corresponding to hsp70 transcripts (arrows in Figure 3C) were significantly smaller than with the FISH<FICC procedure (compare with Figure 3A). Similar results were obtained with hsp90α transcripts (data not shown). This may be due to either a diffusion or a digestion of RNAs by nucleases during the FICC procedure.
In conclusion, we found that FISH<FICC is unambiguously the best procedure to simultaneously detect nuclear transcripts and specific transcription factors. This method was also found to be valuable for the co-detection of UBF nucleolar protein and 28S ribosomal RNAs within the nucleoli. One of the major advantages of this procedure is the absence of permeabilization steps before fixation, thus limiting the degradation of RNAs and the possible occurrence of rearrangements in the overall organization of the nucleus. In consequence, this procedure offers the best compromise between gain of sensivity and preservation of the general nuclear organization. In that respect, it is interesting to point out that both procedures reported here can be used for 3D analysis, because dehydration steps can be omitted without any significant decrease in efficiency (data not shown).
Using this original procedure, we have investigated the relative intranuclear distribution of HSF1 foci and hsp70 transcripts. As shown in Figure 3, we found that the nuclear foci enriched in HSF1 transcription factor do not co-localize with hsp70 gene transcription sites. The distribution of HSF1 foci was also investigated with regard to other active genes (submitted for publication). HSF1 nuclear foci are a new stress-induced compartment that may play a key role in cellular response to stress. Recent studies have shown that other transcription factors, such as GATA (Elefanty et al. 1996) or the mineralo- and glucocorticoid receptors (van Steensel et al. 1995, 1996), are distributed in foci in the interphase nucleus. The functional characterization of these sites of accumulations in relation to transcription sites has been investigated in situ using either BrUTP incorporation or co-detection of transcription factors with their target genes by DNA FISH (van Steensel et al. 1995; Elefanty et al. 1996). However, both approaches have some limitations. The resolution of the BrUTP labeling technique is very limited, and co-detection experiments are rather difficult to analyze because of the large number of labeled sites. On the other hand, detection of target genes, although more fully resolved, does not provide any information on the activity of the genes and requires denaturation of the target. In that respect, nuclear transcript detection by FISH is of particular interest for the characterization of nuclear compartments in relation to specific transcription sites, especially when the transcriptional activity must be assessed.
Footnotes
Acknowledgements
Supported by the Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche, by the Groupement de Recherche et d'Etudes sur le Génome (GREG), the Université J. Fourier (Grenoble I), the Commission of the European Community (Human Genome Analysis contract GENO-CT91-0029 and Human Capital and Mobility CHRX-CT93-0177), and by a fellowship to C.J. from the Ligue Nationale contre le Cancer.
We thank Drs E. Hickey (Reno, NE), R.I. Morimoto (Evanston, IL), and F. Amblard (Grenoble, France) for kindly providing us with the cells, probes, and antibodies used in this study. We are very grateful to F. Zennaf for excellent technical assistance.