
Abstract
We describe the simultaneous localization of DNA sequences in cell and chromosome preparations by means of differently fluorochrome-labeled (AMCA, FITC, TRITC) tyramides using the catalyzed reporter deposition (CARD) procedure. For this purpose, repeated as well as single-copy DNA probes were labeled with biotin, digoxigenin, and FITC, hybridized, and visualized with three different cytochemical detection systems based on horseradish peroxidase conjugates. These were sequentially applied to interphase nuclei and metaphase chromosomes at low concentrations to prevent crossreaction and nonspecific background. In situ localized peroxidase activity was visualized by the deposition of fluorochrome-labeled tyramide molecules. To allow specific deposition of a second and a third tyramide conjugate for multiple-target fluorescence in situ hybridization (FISH), remaining peroxidase activity was always completely inactivated by a mild acid treatment before application of the next peroxidase conjugate. The CARD reactions were optimized for maximal signal-to-noise ratio and discrete localization by tuning reaction time, H2O2, and tyramide concentrations. For both repeated and single-copy DNA targets, high FISH signal intensities were obtained, providing improvement of sensitivity over conventional indirect detection systems. In addition, the fluorescence CARD detection system proved to be highly efficient and easy to implement in multiple-labeling studies, such as reported here for FISH.
Keywords
The latter method, introduced by Bobrow et al. (1989), is based on the deposition of labeled tyramide molecules by peroxidase activity. It has been established that the highly reactive intermediates of this reaction will bind to tyrosine moieties of proteins present in the immediate vicinity of the peroxidase binding site. Because horseradish peroxidase conjugates are commercially available for use in many biochemical assays, the CARD system has been easily implemented not only in immunocytochemistry (Adams et al. 1992; Berghorn et al. 1994; Merz et al. 1995) but also in DNA and RNA detection procedures using FISH (Kerstens et al. 1995; Raap et al. 1995; Macechko et al. 1997; Schmidt et al. 1997). Despite the high sensitivity that could be obtained in these single-target FISH experiments, the discreteness of FISH signals was often limited. Furthermore, no efficient procedure is thus far available for the simultaneous detection of multiple targets.
Here we describe the use of AMCA-, FITC-, and TRITC-labeled tyramides (TSA direct; NEN Life Science Products, Boston, MA) in sensitive multiple-target FISH procedures, enabling the detection of both repeated and single-copy DNA sequences in interphase cells and metaphase spreads (see also Speel et al. 1996). For this purpose, up to three differently labeled (biotin, digoxigenin, FITC) DNA probes were detected in sequence with cytochemical detection systems containing peroxidase conjugates. Each probe was visualized by a peroxidase–tyramide reaction with mild acid treatment between the individual detection steps. This approach for peroxidase inactivation, which we had already utilized effectively for brightfield visualization of multiple DNA sequences in situ (Speel et al. 1994), proved to be an efficient treatment to combine multiple tyramide depositions without crossreaction on the same biological sample. Moreover, the individual peroxidase–tyramide reactions used in this study were optimized for discreteness of nucleic acid localization.
Materials and Methods
Cell Samples
Metaphase spreads were prepared from human peripheral blood lymphocytes by phytohemagglutinin stimulation, hypotonic spreading, and fixation in methanol:acetic acid (3:1, v/v) according to standard procedures. A single-cell suspension of the human transitional cell carcinoma line T24 (DNA index 1.6; trisomic for the centromeres of chromosome 1 and 7; tetrasomic for the centromere of chromosome 17 and the telomere of chromosome 1p) was made by trypsinization of cultured cells, followed by harvesting and fixation in 70% ethanol as described by Hopman et al. (1988). A cell suspension of a human urinary bladder carcinoma with a DNA index of 1.99 and exhibiting an imbalance between the chromosome 11 centromere (three copies) and 11p15 (two copies) [TCC case number 9 (Voorter et al. 1996)] was prepared as described by Hopman et al. (1991).
Pretreatment of Cell Samples
Pretreatment of metaphase spreads with RNAse A and pepsin, followed by postfixation in 1% formaldehyde in PBS (0.15 M NaCl, 10 mM sodium phosphate, pH 7.2) has been described previously (Speel et al. 1992).
Removal of the cytoplasm of T24 cells to improve DNA probe and conjugate penetration was performed as described earlier (Speel et al. 1992). Ten μl of the TCC 9 cell suspension was proteolytically digested with 100 μg/ml pepsin (P-7000; Sigma, St Louis, MO) in 0.01 N HCl for 20 min at 37C to improve DNA probe penetration. The resulting nuclei were neutralized to pH 7.0 by addition of TAE buffer (200 mM Tris-acetate, 50 mM EDTA, pH 8.0) and cytospun at 110 × g for 5 min onto poly-
DNA Probes and Probe Labeling
DNA probes specific for the (sub)centromeric regions of human chromosomes 1 (pUC 1.77), 7 (p7t1), 8 (D8Z2), 11 (pLC11A), 17 (p17H8), the telomeric region of chromosome 1 (p1–79), the 5S rDNA repeat on chromosome 1q42–43 (pH5SB), chromosome 11p15 and 11q23 (40-
FISH Procedures
The labeled (sub)centromere, telomere, cosmid, and plasmid probes were hybridized in single- and multiple-target FISH procedures on metaphase spreads, T24, and TCC 9 cells as described earlier (Speel et al. 1992, 1993, 1994). The 5S rDNA probe had to be hybridized under conditions similar to those used for the cosmid probe due to the presence of an ALU sequence in the 2.3-
Cytochemical Probe Detection
After these washing steps, the slides were preincubated with 4 × SSC, pH 7.0, containing 5% nonfat dry milk (Buffer B) for 10 min at 37C to reduce background staining, followed by dipping in Buffer A. For all the detection procedures, the avidin conjugates were diluted in Buffer B and all the antibody conjugates were diluted in PBS containing 0.05% Tween 20 (Buffer C) and 2% normal goat serum (NGS). After each incubation step of 30 min at 37C, the slides were rinsed twice in Buffer A (avidin conjugates) or Buffer C (antibody conjugates) for 5 min at RT.
Biotin-labeled probes were detected with horseradish peroxidase-conjugated avidin (AvPO, 1:200; DAKO, Glostrup, Denmark) and, optionally, with subsequent layers of biotinylated goat anti-avidin (BioGAA, 1:200; Vector, Brunschwig Chemie, Amsterdam, The Netherlands) and AvPO, or with mouse anti-biotin (MABio, 1:400; DAKO) and horseradish peroxidase-conjugated goat anti-mouse IgG (GAMPO, 1:200; DAKO). For visualization with the alkaline phosphatase–Fast Red reaction, probes were detected with alkaline phosphatase-conjugated avidin (AvAPase, 1:50; DAKO). Digoxigenin-labeled probes were detected with horseradish peroxidase-conjugated sheep anti-digoxigenin Fab fragments (ShADigPO, 1:200; Boehringer), used only for the detection of the (sub)centromeric probes, or with mouse anti-digoxin (MADig, 1:20000; Sigma) and GAMPO. FITC-labeled probes were detected with horseradish peroxidase-conjugated anti-FITC (AFITCPO, 1:2000; NEN Life Science), used only for the detection of the (sub)centromeric probes, or with rabbit anti-FITC (RAFITC, 1:2000; DAKO) and horseradish peroxidase-conjugated swine anti-rabbit IgG (SWARPO, 1:200; DAKO).
Multiple-target detection was carried out essentially as described earlier (Speel et al. 1994). In brief, the procedure involved the following steps: (a) biotin-labeled probe detection; (b) peroxidase reaction with the first fluorochrome-labeled tyramide (see below); (c) inactivation of peroxidase rest activity with 0.01 N HCl for 10 min at RT; (d) digoxigenin-labeled probe detection; (e) peroxidase reaction with the second fluorochrome-labeled tyramide (see below); (f) inactivation as in c; (g) FITC-labeled probe detection; and (h) peroxidase reaction with the third fluorochrome-labeled tyramide (see below).
The largest DNA targets were always detected last in sequence, which occasionally involved a change in the sequence of probe detection. In all cases, control experiments with fluorochrome-conjugated avidin or fluorochrome-conjugated antibody molecules were performed to check the specificity of the observed peroxidase–tyramide staining patterns. In some cases the digoxigenin-labeled probes were detected by subsequent incubations with TRITC-conjugated sheep anti-digoxigenin Fab fragments (ShADigTRITC, 1:100; Boehringer)/FITC-conjugated avidin (AvFITC, 1:500; Vector, Burlingame, CA), BioGAA and AvFITC, or by a sequential application of AMCA-conjugated avidin (AvAMCA, 1:100; Vector), BioGAA, AvAMCA, MADig (1:2000), and TRITC-conjugated rabbit anti-mouse IgG (RAMTRITC, 1:100; DAKO).
Enzyme Cytochemistry
Peroxidase Cytochemistry with Fluorochrome-labeled Tyramides (CARD Amplification). After cytochemical detection of the probes, peroxidase detection was performed by addition of different dilutions of hydroxycoumarin (AMCA)-, carboxyfluorescein (FITC)-, or rhodamine (TRITC)-labeled tyramide stock solutions (1 mg/ml; kindly provided by NEN Life Science) in 100% ethanol (AMCA–tyramide) or PBS (FITC- and TRITC–tyramides) to 1 × Amplification Diluent (NEN Life Science) or PBS containing 0.1 M imidazole, pH 7.6, and 0.001% H2O2, and application of this reaction mixture to the slides (50 μl under a coverslip) for 5–15 min at 37C. The slides were washed two times for 5 min with Buffer C. Currently, the fluorochrome-labeled tyramides are commercially available in TSA (tyramide signal amplification) kits from NEN Life Science.
Alkaline Phosphatase Cytochemistry with Fast Red TR. The alkaline phosphatase–Fast Red reaction was performed as described earlier (Speel et al. 1992). In brief, 1 mg naphthol ASMX phosphate (Sigma) in 250 μl 0.2 M Tris-HCl buffer, pH 8.5, containing 10 mM MgCl2 and 5 mg Fast Red TR salt (Sigma) in 750 μl buffer, were subsequently added to 4 ml buffer with 5% polyvinyl alcohol (PVA,
Embedding and Microscopy
Slides were embedded in 90% glycerol/0.02 M Tris-HCl, pH 8.0, containing 2% 1,4-di-azobicyclo-(2,2,2,)-octane (DABCO; Sigma) and, optionally, 0.5 μg/ml 4′,6-diamidino-2-phenyl indole (DAPI; Sigma), propidium iodide (PI; Sigma), or YOYO (Molecular Probes; Eugene, OR). Slides were analyzed using a Leica DM fluorescence microscope with appropriate filter sets for AMCA, FITC, and TRITC, coupled to a CCD camera and image processing system (MetaSystems; Heidelberg, Germany). Images were recorded using a x63 or x100 objective and corrected for background. It should be emphasized here that although a CCD imaging system was used, the FISH signals described in this report were clearly visible through the microscope.
Results
A multiple-target FISH detection procedure was developed applying multiple enhancement steps with the CARD amplification system. For this purpose, combinations of differently fluorochrome-labeled tyramides (TSA direct) were tested in several cell systems, including interphase preparations of T24 and TCC9 tumor cells and metaphase spreads of normal human peripheral blood lymphocytes. The individual enzyme reactions had been optimized first in single-target FISH procedures before performance of the multiple-target FISH procedures.
Single-target FISH
Discrete in situ localization of different DNA targets using the CARD system as the final detection step could be achieved by optimizing conjugate dilutions, H2O2, and fluorochrome-labeled tyramide concentrations in the reaction medium, and incubation time (5–15 min). Optimal and accurate localization of hybridized (sub)centromeric probes was achieved by using the following:
Standard probe concentrations of 0.4 ng/μl.
One or two detection layers of conjugates, diluted two- to tenfold times more than applied for conventional enzyme cytochemical detection (see Materials and Methods).
1:250, 1:250, and 1:1000 dilutions of 1 mg/ml stock solutions of, respectively, AMCA–, FITC–, and TRITC–tyramides in 1 × Amplification Diluent or PBS containing 0.1 M imidazole, pH 7.6, and 0.001% H2O2 for tyramide deposition.
An enzyme reaction of 5 min at 37C.
Figures 1A–1C show the results of the detection of a biotinylated chromosome 1 probe with AvPO and the three different CARD reactions in T24 cells. All cells display three copies of the chromosome 1 centromere, with accurately localized FISH signals of high signal intensities. The signal intensities were considerably higher than those obtained with conventional, indirect detection methods, enabling a two- to tenfold reduction of CCD recording times while still preserving a higher intensity of FISH signals in most cases over conventional generated spots (for comparison see Figures 1I–1J and 1L–1M). TRITC–tyramide depositions provided the highest sensitivity. In comparison with the produced TRITC–tyramide signals (Figure 1C), similarly intense FISH signals could be generated by the alkaline phosphatase–Fast Red reaction, until now one of the few enzyme reactions producing an accurately localizing fluorescent precipitate. For this purpose, standard dilutions of detection conjugates were used and an enzyme reaction time of 5 min (Figure 1D).
Distinct visualization of smaller DNA targets (1p36 repeat and 5S rDNA repeat, both several 100
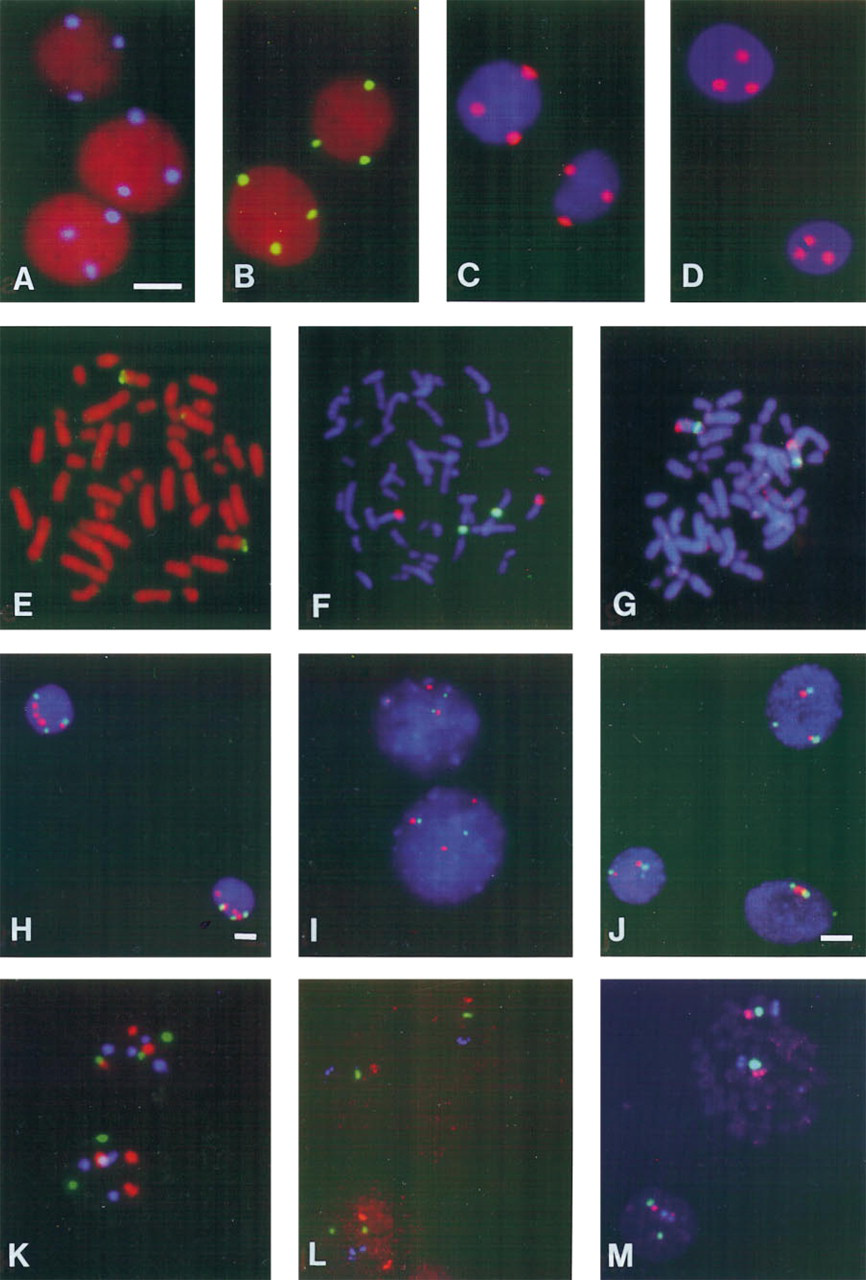
FISH results after CARD (
Double-target FISH
Double-target FISH was performed by hybridizing a biotinylated and a digoxigenin-labeled probe simultaneously on the same metaphase spread or interphase cell preparation and combining the TRITC–tyramide deposition with either the FITC– or AMCA–tyramide deposition reaction for detection. In this way, two copies of the centromeres for chromosomes 1 (FITC–tyramide) and 8 (TRITC–tyramide) could be visualized efficiently in the same metaphase spreads (Figure 1F), while the two chromosome 8 centromeric regions (FITC–tyramide) were detected together with the 15.8-
To determine the effect of the CARD amplification procedure on the number of finally observed FISH signals per nucleus, this procedure was compared with conventional detection on TCC 9 cells, hybridized with the chromosome 11 centromere and 11p15 cosmid probe (e.g., see Figures 1I–1J). For this purpose, 100 intact, nonoverlapping cells were evaluated for both DNA probes. With conventional detection, the main 11c/11p15 ratios were 3/2 (76% of the cells) and 2/2 (12% of the cells), whereas after CARD detection 72% of the cells showed the 3/2 ratio and 14% a ratio of 2/2. The efficiency of DNA target detection is therefore not influenced by the use of CARD detection systems, whereas in the latter case spot counting was much easier due to the high intensity of the FISH signals.
Triple-target FISH
The AMCA–, FITC–, and TRITC–tyramide deposition reactions were combined in triple-target FISH procedures for simultaneous visualization of the centromeres of chromosomes 1 (three copies, FITC), 7 (three copies, TRITC) and 17 (four copies, AMCA) in T24 cells (no counterstaining applied; Figure 1K). As already described for the double-target FISH procedure, mild acid treatment was essential for inactivation of peroxidase rest activities after each CARD reaction. Another example shows the probes for 5S rDNA, the chromosome 1 centromere, and 1p36 on metaphase spreads, detected with AMCA–, FITC–, and TRITC–tyramides, respectively (Figure 1M). In this case, a high detection efficiency could also be achieved with much higher signal intensities than after conventional detection (Figure 1L).
Discussion
In the present study we describe the further optimization and application of the new CARD amplification method for use in multiple-target FISH procedures. A combination of three DNA probes, haptenized with either biotin, digoxigenin, or FITC and hybridized simultaneously to interphase or metaphase preparations, could be cytochemically detected by sequential application of three unrelated, peroxidase-conjugated detection systems, each directly followed by the peroxidase-mediated deposition of blue AMCA-, green FITC-, or red TRITC-labeled tyramides (TSA direct). In this way, three different DNA sequences (repeated as well as single-copy targets) could be localized simultaneously with high sensitivity and efficiency for immediate analysis by fluorescence microscopy. The entire detection procedure, including peroxidase inactivation steps (see below), could be carried out within 3 hr, which was only slightly longer than the conventional detection procedure.
The CARD detection protocol using standard probe concentrations was optimized for maximal signal-to-noise ratio and discreteness of signal localization by adjusting the number of cytochemical detection layers, the dilution of the reagents, the H2O2, and the tyramide concentrations in the reaction medium, as well as the incubation time of the enzyme reaction. When DNA probes containing repetitive elements, such as Alu sequences, were used, a high excess of competitor DNA was a prerequisite to prevent a reduction of the signal-to-noise ratio caused by the CARD amplification reaction. This was also essential for using even more complex probes, such as Alu- or DOP-PCR-amplified sequences, and sometimes needed, in addition, decreasing probe concentrations to obtain acceptable signal-to-noise ratios (data not shown).
In general, detection conjugates could be diluted two- to tenfold further than for conventional detection. Concentrations of AMCA- and FITC-labeled tyramides were found to be optimal at a dilution of 1:250, whereas TRITC–tyramide proved to be the most sensitive, as has been reported similarly by Raap et al. (1995), at a dilution of 1:1000, using in all cases stock solutions of 1 mg/ml. Optimal enzyme reactions used 0.001% H2O2 and were performed for 5–15 min at 37C, depending on the DNA target to be detected. The centromere-specific probes could be detected efficiently with one detection layer in combination with a 5-min CARD reaction. For low-copy repeated sequences (1p36 and 5S rDNA) and single-copy sequences, minimally two detection layers and a 5-min enzyme reaction were required to ensure proper visualization of all hybridization sites. Detection of these sequences with only one layer often resulted in asynchronous amplification of the FISH signals to be visualized or a decrease in the detected number of signals per nucleus. This may be caused by too low concentrations of peroxidase molecules at the target site, below the critical number that is required to produce enough reaction product to visualize it in the microscope. This phenomenon is intrinsic to enzyme reactions, as has been described for the alkaline phosphatase–Fast Red reaction (Speel et al. 1992). In contrast, others have shown the localization of a cosmid probe with one detection layer (Van Gijlswijk et al. 1996), although in this case a 30-min enzyme reaction was needed in combination with the use of diffusion-limiting reagents, such as polyvinyl alcohol or dextran sulfate. Our experiments, however, demonstrated that the use of multiple detection layers together with short enzyme reactions (5–15 min) are preferred to localize small DNA probes efficiently. Fine tuning of the reaction time is recommended when several slides are available. On the other hand, two CARD reactions can be performed consecutively when the signal intensity is insufficient after one amplification round.
Although we have not yet quantified the FISH signal intensities after CARD amplification and compared the results with conventionally developed signals, in most cases CCD recordings of these images were two- to tenfold shorter than for conventional detection procedures. For example, the exposure times for Figure 1 were 2.6, 0.44, and 0.24 sec for AMCA, FITC, and TRITC CARD images, respectively, whereas they were 5.0, 1.92, and 3.8 sec for the conventional images in Figure 1L. This suggests an amplification factor of at least two- to tenfold with acceptable resolution of the CARD signals.
It is now generally assumed that intensification of FISH signals observed after CARD amplification results from the binding of peroxidase-generated tyramide radicals to tyrosine moieties present in the (fixed) cell preparations. It was to be expected that the use of cytochemical blocking steps, such as nonfat dry milk, increases the binding sites for tyramide radicals. However, experiments comparing different blocking reagents (nonfat dry milk, 5% normal goat serum, and 3% BSA) with no blocking on T24 cells resulted only in slightly different staining intensities, suggesting that application of blocking steps is not essential for optimal signal generation.
The utilization of multiple CARD reactions on one sample requires efficient peroxidase inactivation steps in between two subsequent enzyme reaction steps. The 0.01 N HCl inactivation procedure, which we also used for multiple-target in situ hybridization combined with brightfield microscopy (Speel et al. 1994), appeared to affect neither the final FISH results nor the cell morphology. Therefore, further extension of labels can be explored by utilizing other fluorochromes coupled to tyramine, such as the Cyanine series of dyes and Texas Red, or by using mixtures of multiple fluorochrome-labeled tyramides in one peroxidase reaction. Preliminary experiments have already enabled the detection of three DNA targets together with DNA counterstaining (data not shown). However, the total number of targets to be detected with different CARD reactions will ultimately depend on several factors, including the number of probe labels that can be detected with unrelated detection systems, the stability of fluorochromes during peroxidase inactivation, and the number of fluorochromes that can be accurately separated. In this respect, one should also consider combinations of CARD reactions with alkaline phosphatase reactions resulting in fluorescent precipitates, such as naphthol ASMX phosphate/Fast Red and ELF-97 phosphate (Speel et al. 1995).
We conclude that the fluorescence CARD detection of multiple DNA targets in situ can be applied efficiently and rapidly on different cell preparations, combining the high sensitivity of the CARD system with only minor loss of resolution. This method will be of great value to further increase the detection sensitivity of FISH on different biological materials. Moreover, it will facilitate the counting of FISH signals, which can be advantageous for evaluation of chromosomal aberrations in cytological and histological material.
Footnotes
Acknowledgements
Supported by the Netherlands Organization for Scientific Research NWO, grant no. 900–534–102, and by the Dutch Cancer Foundation, grant no. IKL 92–07.