
Abstract
The heparin affin regulatory peptide (HARP) growth factor, also known as pleiotrophin, is a developmentally regulated protein that displays biological functions during cell growth and differentiation. To study the physiological role of this protein, we investigated the cellular distribution of HARP mRNA and protein in the resting human mammary gland. In situ hybridization histochemistry revealed that HARP mRNA was localized in alveolar myoepithelial cells, whereas alveolar epithelial cells were negative. In the stroma, HARP mRNA was localized in endothelial cells and smooth muscle cells of blood vessels. Interestingly, HARP protein and mRNA were not always co-localized. HARP protein immunocytochemistry staining was observed in an area including both alveolar myoepithelial and epithelial cells, although epithelial cells do not express HARP transcript. In contrast, the distribution of HARP protein is parallel to that of HARP mRNA in endothelial and vascular smooth muscle cells. In the light of these results, the putative role of HARP in controlling the proliferation and/or differentiation of the different mammary cell types is proposed and discussed.
Keywords
Distribution of HARP mRNA using in situ hybridization in embryonic as well as adult rat tissues has been previously studied (Vanderwinden et al. 1992). Its expression distribution during embryonic and postnatal development suggests important functions in tissue growth and differentiation. Moreover, the presence of HARP transcripts in adult tissues, including meninges, iris, testis, and uterus, also indicates a physiological role during adulthood. To further investigate the potential role of HARP in adult and normal tissues, we have focused our study on the cellular localization of HARP mRNA and immunoreactive HARP polypeptide in normal human mammary gland (NHMG). Our results provide new information that raises new suggestions for the physiological function of HARP in this organ.
Materials and Methods
Tissue Samples
Human normal breast tissue biopsy specimens were obtained immediately after surgery from the Department of Pathology of the Institut Curie (Paris, France) and the Henri Mondor hospital (Créteil, France). They were taken from areas opposite to invasive adenocarcinomas (n = 11) and from reduction mammoplasties (n = 6). Frozen tissue sections 5 μm thick were applied to SuperFrost/Plus glass slides (Kindler; Freiburg, Germany) and fixed for 15 min in acetone at 4C for immunocytochemistry. For in situ hybridization, sections were fixed in 4% neutral-buffered formalin at room temperature (RT) for 15 min and dehydrated through graded ethanols. Fixed sections were then stored at −20C until used.
Production of Anti-HARP Antibody
Polyclonal antibodies against HARP were raised in rabbits as follows: One hundred μg of purified human recombinant HARP diluted in PBS containing 5 mg/ml of heparin was emulsified with Freund's complete adjuvant (1:1) and injected
Western Blotting Assay
Frozen biopsy specimens of human mammary glands were disrupted in 20 mM Hepes, pH 7.4, 1 μg/ml aprotinin, leupeptin, and pepstatin (Sigma; St Louis, MO), 0.1 mM PMSF, 3 mM EDTA (lysis buffer) containing 2 M NaCl using an Ultra-Turrax T25 (Jankle & Kunkel; Staufen, Germany). The clarified supernatant from the centrifugation step was diluted fivefold with lysis buffer and incubated overnight at 4C with heparin–Sepharose CL-6B beads (Pharmacia) on a rotating rack. The beads were washed twice with 20 mM Hepes, pH 7.4, 0.5 M NaCl, and once with 20 mM Hepes pH 7.4. Heparin–Sepharose-bound molecules were then eluted with electrophoresis sample buffer (50 mM Tris-HCl, pH 6.8, 10% glycerol, 0.02% bromophenol blue, 2% SDS, and 5% β-mercaptoethanol) at 95C for 5 min.
Samples were loaded and run on an SDS-polyacrylamide gel before transfer to Immobilon-P membrane (Millipore; Saint-Quentin–Yvelines, France) in 10 mM CAPS, pH 11, 10% MeOH using a Sartorius electrophoretic transfer system. Nonspecific binding was prevented by incubating the membrane in PBS-T [phosphate-buffered saline (PBS), 0.2% Tween-20] plus 3% gelatin. The membrane was then incubated overnight at 4C with rabbit anti-HARP Ig at 1:500 dilution in PBS-T plus 1.5% normal goat serum. After washes in PBS-T, bound antibodies were visualized with peroxidase-conjugated goat anti-rabbit IgG (Sanofi Diagnostic Pasteur; Marnes la Coquette, France) and ECL (Amersham Life Science; Poole, UK) as substrate. The membrane was then processed for autoradiography by exposure to Kodak X-Omat film for 3–30 min.
Immunocytochemistry
Tissue sections were rehydrated in PBS. Nonspecific binding of antibodies was prevented by incubating the slides with PBS-T containing 1.5% normal goat serum and 1% gelatin (Buffer A) at RT for 30 min. After a blocking step with an avidin–biotin blocking kit (Vector Laboratories; Burlingame, CA), sections were incubated for 30 min at 37C in Buffer A containing 5 μg/ml of affinity-purified anti-HARP Ig. After two washes in PBS-T, a second incubation for 30 min at RT using biotinylated anti-rabbit IgG (Vector Laboratories) was carried out at a dilution of 1:200 in Buffer A. After two washes in PBS-T, sections were incubated for 30 min at RT with an avidin–biotin–alkaline phosphatase complex and red coloration and fluorescent staining was obtained using the Vector red substrate (Vector Laboratories). Specific staining controls included omission of the first antibody, incubation with the first antibody supplemented with a 50-fold molar excess of FGF-2 or EGF, and replacement of the purified anti-HARP Ig with the HARP-HiTrap affinity-chromatography unbound Ig used at 5 μg/ml. Immunocytochemical staining with mouse antiserum against human Factor VIII-related antigen (Dako; Glostrup, Denmark) was performed in the same way as that used for HARP Ig, except that the second incubation was performed with an alkaline phosphatase (AP)-conjugated anti-mouse IgG (Dako). All sections were counterstained with Harris's hematoxylin before mounting in Aquamount (BDH; Nottingham, UK). Sections were then viewed in a Zeiss microscope and photographed on Kodak 100 ASA film.
In Situ Hybridization
All reagents were purchased from Boehringer Mannheim (Mannheim, Germany) except those for the polymerase chain reaction (PCR), which were from Promega (Madison, WI). Detection of HARP mRNA was undertaken using a digoxigenin (DIG)-11-dUTP-labeled probe according to Caruelle (unpublished procedure). Sense and anti-sense DIG-11-dUTP-labeled probes were prepared as follows. A linear 1200-bp cDNA containing the open reading frame of the HARP gene was subjected to 35 cycles in a standard PCR using 1 μM of sense 5′-GAAAATTTGCAGCTGCCTT-3′ and anti-sense 5′-CTTCTCCTGTTTCTTGCCT-3′ primer (Eurogentec; Seraing, Belgium) and 0.2 mM dATP, dCTP, dGTP, dTTP; 50 mM KCl, 10 mM Tris-HCl, pH 8.3; 1.5 mM MgCl2; 0.01% gelatin; 1.25 U of Taq polymerase. The 464-
Sections were rehydrated, washed in 10 mM Tris-HCl, pH 7.5, incubated for 30 min at 37C with 10 μg/ml of proteinase K in 20 mM Tris-HCl, pH 7.5, 2 mM CaCl2, and postfixed in 4% neutral-buffered formalin for 15 min at RT. After washing in PBS and 4 × SSC, sections were prehybridized for 4 hr at 37C with hybridization buffer in a humid chamber. Hybridization was performed overnight at 37C with 25 μl/section of DIG-labeled sense or anti-sense probe. Posthybridization washes were for 30 min each as follows: 2 × SSC, RT; 1 × SSC, RT; 0.5 × SSC, 37C; and finally 0.5 × SSC, RT. Sections were then preincubated in 3% normal sheep serum and 1% Boehringer blocking reagent (BBR) in 100 mM Tris-HCl, pH 7.4, 150 mM NaCl (7.4 AP buffer) for 45 min at RT, followed by an incubation with 1:200 diluted AP-conjugated anti-digoxigenin in 7.4 AP buffer containing 1% BBR, 0.3% Triton X-100 for 90 min at RT. The sections were washed twice in 7.4 AP buffer and once in 100 mM Tris-HCl, pH 9.5, 150 mM NaCl, 50 mM MgCl2 (9.5 AP buffer). Color was developed by incubation in 9.5 AP buffer containing 0.33 mg/ml nitroblue tetrazolium chloride (NBT) and 0.17 mg/ml of 5-bromo-1-chloro-3-indolyl phosphate (BCIP) for 2–3 hr at RT. Reaction was achieved by washing the sections in 7.4 AP buffer. Sections were mounted in Aquamount (BDH), then viewed in a Zeiss microscope and photographed on Kodak 100 ASA film.
Results
Cellular Distribution of HARP mRNA in NHMG
In situ hybridization histochemistry showed that HARP mRNA staining was entirely confined to alveolar myoepithelial cells surrounding the glandular structure, whereas alveolar epithelial cells did not display positive HARP mRNA hybridization (Figures 1C and 1D). Negative control sections incubated with sense probe showed no staining, demonstrating the specificity of the DIG-labeled probe system (Figure 1B). In addition, HARP transcripts were also strongly expressed in blood vessels present in the stroma of NHMG. More precisely, and as shown in Figure 1E, the staining occurred in capillaries, since adjacent serial section of the same capillary was positively stained by an anti-factor VIII-related antigen serum (Figure 1F) and negatively by an anti-α-smooth actin-related antigen serum (not shown), thus demonstrating that endothelial cells expressed HARP mRNA. Moreover, strong staining was also seen in a cross-section of an arteriole, indicating that HARP mRNA was synthesized by vascular smooth muscle cells (Figure 1G).
Validation of Anti-HARP Antibodies and Immunochemical Staining
Specificity of the rabbit affinity-purified antibodies was confirmed by Western blotting analysis of purified HARP, MK, FGF-2, and EGF proteins. Figure 2 shows that affinity-purified Ig bound to HARP but failed to recognize MK, FGF-2, and EGF molecules, indicating no crossreactivity. Western blotting of heparin-purified cell extracts of human normal mammary gland with anti-HARP Ig produced only a unique immunoreactive signal that co-migrated with the 18-kD HARP protein.
Immunocytochemical Staining of HARP Protein in NHMG
Immunocytochemical staining using specific rabbit antibodies to HARP protein showed this growth factor to be localized in a region that included both alveolar epithelial and myoepithelial cells (Figures 1H and 1I). In the stroma, the matrix compartment was clearly negative. No staining was observed when negative control sections were incubated either with affinity chromatography-unbound Ig (Figure 1J) or with PBST in replacement of the first antibody (not shown). Moreover, no modification of labeling was seen when the slides were incubated with the first antibody supplemented with a 50-fold molar excess of FGF-2 or EGF (not shown). In agreement with the mRNA localization, immunostaining was observed in capillaries (Figure 1K) and arterioles (Figure 1L).
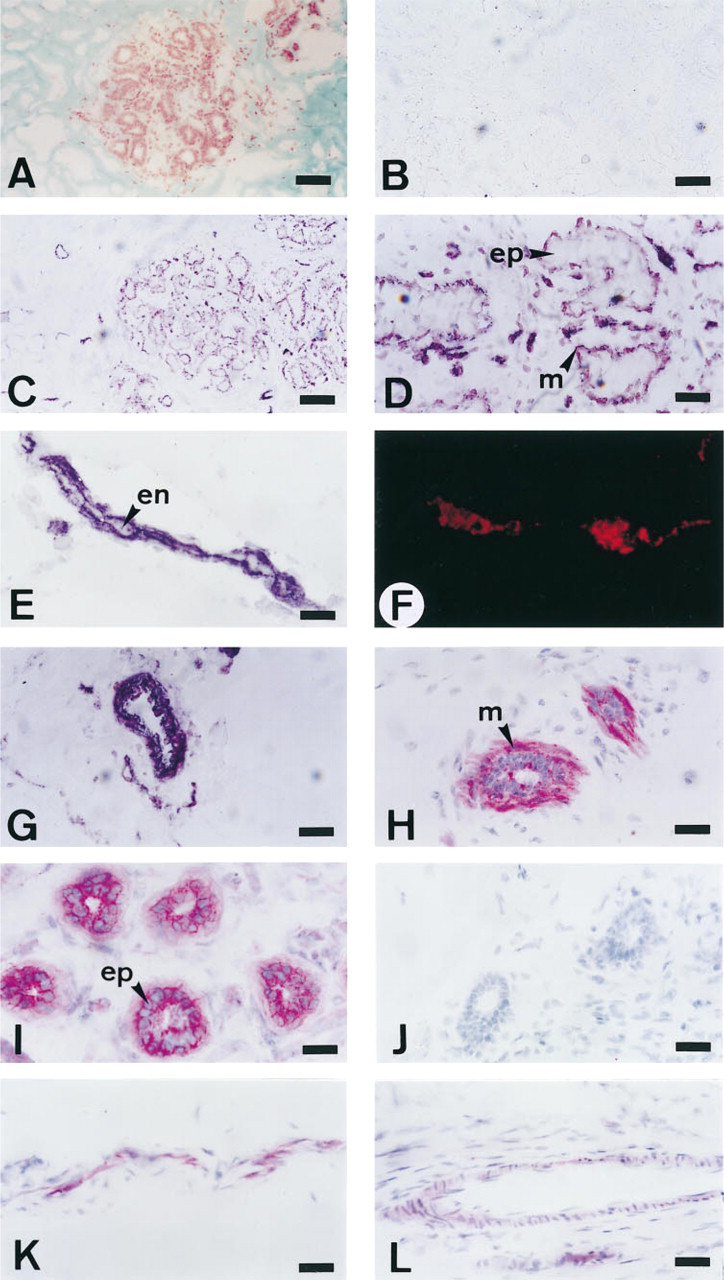
Cellular localization of HARP mRNA (
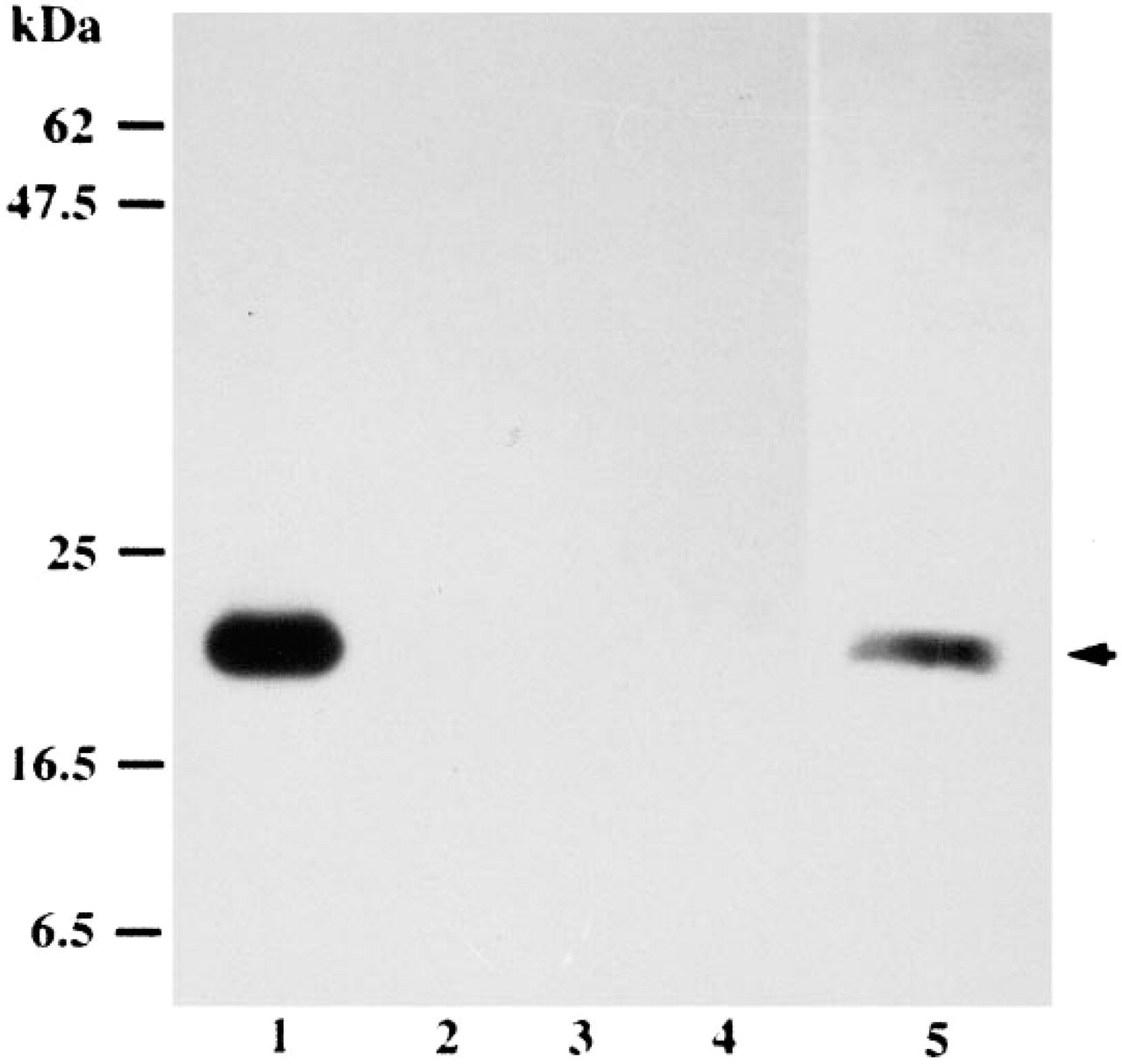
Western blotting assays. HARP (Lane 1), FGF-2 (Lane 2), MK (Lane 3), EGF (Lane 4), and heparin-purified extracts of human mammary gland (Lane 5) were fractionated by SDS-PAGE before transfer to Immobilon-P, as described in Materials and Methods. The immunoreactive component is visualized as a single stained 18-kD band (arrow).
Discussion
The strong expression of HARP mRNA and protein originally observed during postnatal development of rat brain suggested that HARP may play important functions in the growth and differentiation of neuronal cells (Rauvala 1989; Merenmies and Rauvala 1990). However, the tissue distribution of HARP mRNA using in situ hybridization in embryonic and adult rat tissues (Vanderwinden et al. 1992) indicated that HARP is also present in non-neuronal tissues. HARP mRNA has been also detected in various cell lines originally obtained from breast cancer (T47 Dco, MDA-MB231, MDA-MB361, or Hs-578T), prostate cancer (PC-3), ovarian carcinoma (A1827, PA-1) (Fang et al. 1992), and in normal human intestinal smooth muscle cells (HISM) (Li et al. 1992), suggesting that HARP could be found in vivo in the corresponding tissues and was therefore more broadly distributed than previously reported. At this time, the role of HARP remains obscure and little is known about the physiological or pathophysiological functions of this molecule. To contribute to a better understanding of the in vivo physiological function of this molecule, it is essential to know more about the precise tissue localization of both the protein and its mRNA. We therefore focused our investigation on the normal human mammary gland (NHMG) as a necessary step before looking at pathological mammary tissues.
The presence of HARP mRNA in biopsy specimens from both normal and tumoral human mammary gland has been previously described using reverse transcriptase-polymerase chain reaction (RT-PCR) (Garver et al. 1994). However, the transcripts used in this study were isolated from resected human tissues composed of a wide variety of cell types and this technique could not provide any information on the cellular distribution of HARP mRNA. We present here for the first time the localization of both HARP protein and its mRNA in NHMG. We show that in the glandular compartment, HARP protein and its mRNA are mainly localized and synthesized in alveolar myoepithelial cells. Interestingly, HARP protein was detected in areas of alveolar epithelial cells, even though its transcript was not detected. Although the physiological function of HARP in glandular epithelial cells remains unknown, this observation suggests a paracrine mechanism of HARP for these epithelial cells. One possible mechanism for the presence of HARP protein in the vicinity of epithelial cells is that HARP is synthesized and secreted by myoepithelial cells and could diffuse up to the extracellular matrix and/or the cell surface membrane of epithelial cells. Hence, during the normal growth and development of the mammary gland, HARP may be released from this storage site and act via an autocrine or paracrine mechanism on either the myoepithelial cells themselves or the adjacent epithelial cells, stimulating their proliferation and/or differentiation.
HARP mRNA and protein were also detected both in the endothelial and smooth-muscle cells of blood vessels. Several studies have demonstrated that purified human recombinant HARP was mitogenic for capillary endothelial cells and possessed angiogenic activity (Courty et al. 1991; Fang et al. 1992; Laaroubi et al. 1994; Czubayko et al. 1995). In addition, a more recent report demonstrated that the C-terminal part of HARP enhanced plasminogen activator levels and down-regulated PAI-1 expression in bovine aortic endothelial cells (Kojima et al. 1995). Furthermore, it has been shown that HARP belongs to the family of heparin-releasable proteins that are molecules that contribute to regulation of vascular homeostasis and angiogenesis (Novotny et al. 1993). Therefore, although the exact physiological role of HARP localized in blood vessels of the NHMG requires greater clarification, these data support the idea that HARP protein can play a key role in angiogenesis mechanisms that require endothelial cells to migrate, to degrade the extracellular matrix, to proliferate, and to differentiate into capillaries.
Interestingly, it is noteworthy that HARP and fibroblast growth factor-2 (FGF-2) are co-localized in myoepithelial cells and in blood vessels (Gomm et al. 1991; Rudland et al. 1993). The functional meaning of this co-localization remains to be investigated. However, it has been reported that FGF-2 regulates HARP gene expression in vitro (Li et al. 1992; Merenmies 1992) and that MK, which displays a 55% amino acid sequence homology with HARP, stimulated cell proliferation in mandibular mesenchyme from the tooth region when it was added with the FGF-2 molecule (Mitsiadis et al. 1995). Although these observations were from distinct and in vitro models, it is tempting to speculate that in vivo, and especially in the NHMG, HARP and FGF-2 may be acting in parallel by regulating in concert their biological activities.
In conclusion, we have now collected more information on HARP in normal mammary tissues showing that myoepithelial, endothelial, and vascular smooth muscle cells expressed both HARP mRNA and protein. It is noteworthy that this mRNA and protein pattern of localization was observed in biopsies derived from an area opposite to adenocarcinoma (n = 11) as well as from mammoplasties (n = 6). In any event, it now appears of greatest interest to perform the same type of investigation in pathological mammary tissues to evaluate whether this molecule could be a major factor in the malignant progression of breast cancer, as has been previously suggested (Riegel and Wellstein 1994).
Footnotes
Acknowledgements
Supported by grants from the Association pour la Recherche sur le Cancer (no. 6595), the Ministère de l'éducation Nationale (DRED), the Ligue Nationale Contre le Cancer, and Naturalia et Biologia.
We are grateful to Dr Bagot and Dr Charu (Henri Mondor hospital, Créteil, France) and Dr Zafrani and Dr. Sastre (Institut Curie, Paris, France) for providing breast biopsies.