
Abstract
Insulin stimulates glucose transport in rat adipose cells through the translocation of GLUT4 from a poorly defined intracellular compartment to the cell surface. We employed confocal microscopy to determine the in situ localization of GLUT4 relative to vesicle, Golgi, and endosomal proteins in these physiological insulin target cells. Three-dimensional analyses of GLUT4 immunostaining in basal cells revealed an intracellular punctate, patchy distribution both in the perinuclear region and scattered throughout the cytoplasm. VAMP2 closely associates with GLUT4 in many punctate vesicle-like structures. A small fraction of GLUT4 overlaps with TGN38-mannosidase ll, γ-adaptin, and mannose-6-phosphate receptors in the perinuclear region, presumably corresponding to late endosome and trans-Golgi network structures. GLUT4 does not co-localize with transferrin receptors, clathrin, and lgp-120. After insulin treatment, GLUT4 partially redistributes to the cell surface and decreases in the perinuclear area. However, GLUT4 remains co-localized with TGN38-mannosidase ll and γ-adaptin. Therefore, the basal compartment from which GLUT4 is translocated in response to insulin comprises specialized post-endosomal VAMP2-positive vesicles, distinct from the constitutively recycling endosomes. These results are consistent with a kinetic model in which GLUT4 is sequestered through two or more intracellular pools in series.
However, many questions regarding the localization and trafficking pathways of GLUT4 remain unanswered. At present, the nature of the membrane compartment in which GLUT4 is sequestered in the basal state remains poorly defined. Moreover, it is not known whether the GLUT4-containing vesicles belong to an adipose cell adaptation of an endosomal compartment or to a specialized secretory exocytic compartment.
Other than the studies cited above, immunofluorescence at the light microscopic level and colloidal gold immunocytochemistry at the electron microscopic level have focused on the use of plasma membrane lawns (sheets) as an assay for GLUT4 translocation (Robinson et al. 1992; Voldstedlund et al. 1993). This technique is limited to an examination of structures associated with the inner surface of the plasma membrane.
Extensive biochemical studies have been performed in search of other molecules residing in the same vesicles which contain GLUT4. Co-localization studies have shown that the subcellular distributions of endosomal proteins such as transferrin receptors (TfR) and insulin-like growth factor-ll/mannose-6-phosphate receptors (IGFllR/M6PR) overlap with that of GLUT4. However, these studies, using mainly biochemical techniques and cultured cells, have produced contradictory results, ranging from complete overlap between TfR and GLUT4 (Davis et al. 1986; Tanner and Lienhard 1987, 1989), to partial overlap (Hudson et al. 1992, 1993; Laurie et al. 1993; Hanpeter and James 1995; Livingstone et al. 1996), to no overlap at all (Herman et al. 1994). Similarly, subcellular fractionation has suggested various degrees of co-localization between IGFllR/M6PR and GLUT4 (Tanner and Lienhard 1989; Zorzano et al. 1989; Martin et al. 1994; Kandror and Pilch 1994, 1996; Hanpeter and James 1995). At present it is still unclear what proportion of GLUT4 is targeted to the endosomal compartment under basal conditions.
Several studies have suggested that GLUT4-containing vesicles might belong to a regulated secretory pathway, either as unique vesicles or similar to those of the synaptic vesicle compartment. When GLUT4 were overexpressed in cells that have a regulated secretory pathway, they appeared to be targeted to the secretory granules (Hudson et al. 1993). A major 165-kD protein, identified as an aminopeptidase, has been found associated with GLUT4-containing vesicles (Kandror and Pilch 1994; Kandror et al. 1994; Mastick et al. 1994). Vesicle-associated membrane proteins (VAMPs), such as synaptobrevin 2 (VAMP2) and cellubrevin (VAMP3) (Cain et al. 1992; McMahon et al. 1993; Trimble et al. 1993; Volchuk et al. 1994, 1995; Ralston et al. 1994; Timmers et al. 1996), and secretory carrier membrane proteins (SCAMPs) (Laurie et al. 1993; Thoidis et al. 1993) have also been found in GLUT4-containing vesicles using biochemical approaches. These proteins were initially considered to be specific components of the regulated secretory pathway, suggesting common vesicle processing for glucose transporters and secretory proteins. However, the specificity of these proteins has recently been challenged, and therefore they are no longer suitable tools for mapping out the vesicles that move towards the plasma membrane (Grote and Kelly 1996). Finally, biochemical approaches alone can never fully define the very complex and dynamic three-dimensional organization of the compartments through which GLUT4 traffics in the intact cells in response to insulin.
In this study we developed an immunocytochemical approach to determine the whole-cell subcellular distribution of GLUT4 and its co-localization with compartment markers in rat adipose cells in the basal and insulin-treated states. To obtain images that would more accurately reflect the distribution of GLUT4 in different regions of the cell, we employed immunofluorescence confocal microscopy with optical sectioning and three-dimensional reconstruction. Adipose cells are natural target cells for insulin's stimulatory action on glucose transport activity, and therefore we performed overlapping studies using isolated white and brown adipose cells. As an initial step towards understanding the GLUT4 trafficking pathways, our data have led us to conclude that the basal intracellular GLUT4 compartment comprises specialized post-endosomal vesicles containing VAMP2.
Materials and Methods
Preparation and Incubation of Adipose Cells
Male Sprague–Dawley rats (170–200 g, CD strain) from Charles River Breeding Laboratories (Boston, MA) were used. The rats were anesthetized with a mixture of 70% CO2, 30% O2 and sacrificed by decapitation. The epididymal fat pads were removed and white adipose cells were isolated with collagenase digestion as described previously (Weber et al. 1988). Brown adipose tissue was dissected from the interscapular region and brown adipose cells were isolated with a collagenase/DNAse digestion, also as previously described (Omatsu–Kanbe et al. 1996). All incubations were carried out at 37C in a Krebs–Ringer–bicarbonate–HEPES buffer, pH 7.4, containing 10 mM sodium bicarbonate, 30 mM HEPES, 200 nM adenosine (KRBH buffer), and 1% bovine serum albumin (fraction V) (BSA) from Intergen (Purchase, NY). Isolated adipose cells (2–4 × 106 cells/ml) were distributed equally to plastic vials and incubated without or with 700 nM insulin (Eli Lilly; Indianapolis, IN) at 37C for 30 min. Glucose transport activity was assessed by the tracer [U-14C]-glucose uptake method (Foley et al. 1983).
Antibodies
Two antibodies were used to localize GLUT4 and several antibodies were used to localize various cellular “compartment markers.” For GLUT4, we employed an affinity-purified polyclonal rabbit IgG (0.15 μg/ml) kindly provided by Hoffmann–La Roche (Nutley, NJ) and the mouse monoclonal antibody F-27 (1 μg/ml) kindly provided by Dr. P.N. Jorgensen of Novo Nordisk (Bagsvaerd, Denmark). Both antibodies are directed to C-terminal sequences of GLUT4 (of 20 and 14 amino acid residues, respectively) and their specificities were previously demonstrated by immunochemical and immunocytochemical techniques (Holman et al. 1990; Ploug et al. 1993; Voldstedlund et al. 1993).
The following primary antibodies used in our double labeling experiments were generously provided as gifts and have been previously characterized: the affinity-purified rabbit polyclonal anti-VAMP2 antibody LL220 (1 μg/ml) raised against a synthetic peptide corresponding to amino acids 1–16 of rat VAMP2 from Dr. M. Knepper of the NIH (Bethesda, MD) (Sudhof et al. 1989; Gaisano et al. 1994; Nielsen et al. 1995); a rabbit polyclonal anti-M6PR/IGFllR antibody (whole serum used at 1:40 dilution) from Dr. P. Nissley of the NIH (Dahan et al. 1994); and a mouse monoclonal anti-rat TGN38 antibody (1:2 dilution of ascites fluid) (Horn and Banting 1994; Reaves et al. 1996) and a mouse monoclonal anti-mannosidase ll antibody (1:2 dilution of ascites fluid) (Burke et al. 1982), both from Dr. J.P. Luzio and Dr. B. Reaves of Cambridge University (Cambridge, UK). Because the staining patterns observed with anti-TGN38 and anti-mannosidase ll antibodies are very similar, the two MAbs were usually used together (1:1) to obtain a higher signal on immunostaining. Additional antibodies were an affinity-purified rabbit polyclonal anti-γ-adaptin antibody (5 μg/ml) raised against a glutathione-S-transferase fusion protein from Dr. M. Robinson of Cambridge University (Page and Robinson 1995) and the mouse monoclonal anti-lysosomal glycoprotein-120 (lgp-120) antibody Ly1C6 (5 μg/ml) from Dr. I. Mellman of Yale University (New Haven, CT) (Lewis et al. 1985).
The following antibodies and conjugates were from commercially available sources: a rhodamine (Rhd)-conjugated lectin Lens culinaris agglutinin (LCA) (50 μg/ml) from Vector Laboratories (Burlingame, CA); the mouse anti-rat TfR MAb MAB1451 (7 μg/ml) from Chemicon International (Temecula, CA); and the mouse anti-clathrin H (heavy) chain MAb CHC5.9 (10 μg/ml) from ICN Biomedicals (Costa Mesa, CA). Fluorescein isothiocyanate (FITC)- and lissamine rhodamine sulfonyl chloride (LRSC)-conjugated antibodies specific for rabbit or mouse immunoglobulins (Ig), used at 15 μg/ml as secondary antibodies in immunofluorescence experiments, were obtained from Jackson ImmunoResearch (West Grove, PA).
Indirect Immunofluorescence Microscopy
Single immunofluorescence experiments were performed using adipose cells in suspension throughout the following protocol. Isolated white or brown adipose cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences; Ft Washington, PA) in PBS 0.15 M, pH 7.4, for 20 min at room temperature (RT) and then washed three times with PBS. Subsequently the cells were permeabilized and the nonspecific binding sites were blocked with 0.1% saponin (Sigma; St Louis, MO) in PBS containing 1% BSA and 3% normal goat serum for 45 min at RT. The cells were then incubated with the primary antibodies brought to the appropriate dilutions in the same solution for 2 hr at RT. After this time the cells were washed with PBS containing 0.1% saponin to remove the unbound immunoglobulins, incubated for 1 hr at RT with the fluorochrome-conjugated secondary antibodies, and washed again. Cells were mounted on glass slides using Vectashield mounting medium (Vector Laboratories; Burlingame, CA).
In double labeling experiments, pairs of anti-GLUT4 antibodies and the corresponding secondary antibodies were used in conjunction with pairs of anti-compartment marker antibodies and the corresponding secondary antibodies. The procedures were conducted sequentially. The first pair of primary and secondary antibodies was followed by a second pair that was also applied in two steps. For localization of the plasma membrane, fixed cells were first incubated with the rhodamine-labeled lectin LCA for 30 min at 4C, washed, and then permeabilized and incubated with anti-GLUT4 antibodies as just described. For immunolocalization of clathrin, cells were permeabilized with 0.1% Triton X-100 for 15 min at RT. Control experiments were performed in which the specific antibodies were omitted or replaced with nonimmune serum.
Microscopy and Image Analysis
Staining was observed with the Nikon Optiphot 2 fluorescence microscope equipped with a Bio-Rad MRC-600, 1000, or 1024 confocal laser scanning imaging system (CLSM) from Bio-Rad Labs (Hercules, CA). This system utilizes a mixed argon/krypton laser (λ1 = 488 nm, blue line for FITC; λ2 = 568 nm, yellow line for rhodamine) and COMOS/Lasersharp image analysis software. Specimens were viewed at high magnification using planapochromat x 60/1,4 NA and x 100/1,4 NA oil objectives. For each experimental condition, 8–10 images/cell were recorded from at least 10–15 cells. Images were collected sequentially for the two fluorochromes in the double labeling experiments using Kalman averaging at an optical zoom setting of 1 to 2.5 (Pawley 1995). Co-localization was assessed throughout the cell by examination of several merged images and was expressed as “high” when the overlap was present in all focal plans, “partial” when the overlap was present in one to three focal planes, and “absent” when no significant overlap was observed in one focal plane. The degree of co-localization was estimated semiquantitatively by analysis of merged images before processing for illustration. For three-dimensional reconstruction, series of optical sections were collected at 0.5-μm intervals along the Z-axis using the CLSM (Matsumoto 1993). For presentation, images were further enhanced digitally using the Adobe Photoshop 3.0 program from Adobe Systems (Mountain View, CA) and printed with a Kodak 8650 PS digital printer (Eastman Kodak; New Haven, CT).
Results
We have investigated the subcellular localization of GLUT4 and compared it to those of various organelle markers by confocal laser scanning immunofluorescence microscopy. We have used isolated white and brown rat adipose cells in our study because these cells constitute physiological target cells for insulin action. Immunocytochemistry at both the light and the electron microscopic level, using rat adipose cells, is not trivial because of their geometry, size, and high content of lipid, and most of the published results are based on data obtained using cultured cell lines. Despite some technical challenges caused by the high content of lipid, we have found that freshly isolated white and brown adipose cells from the rat are both suitable for immunofluorescence studies.
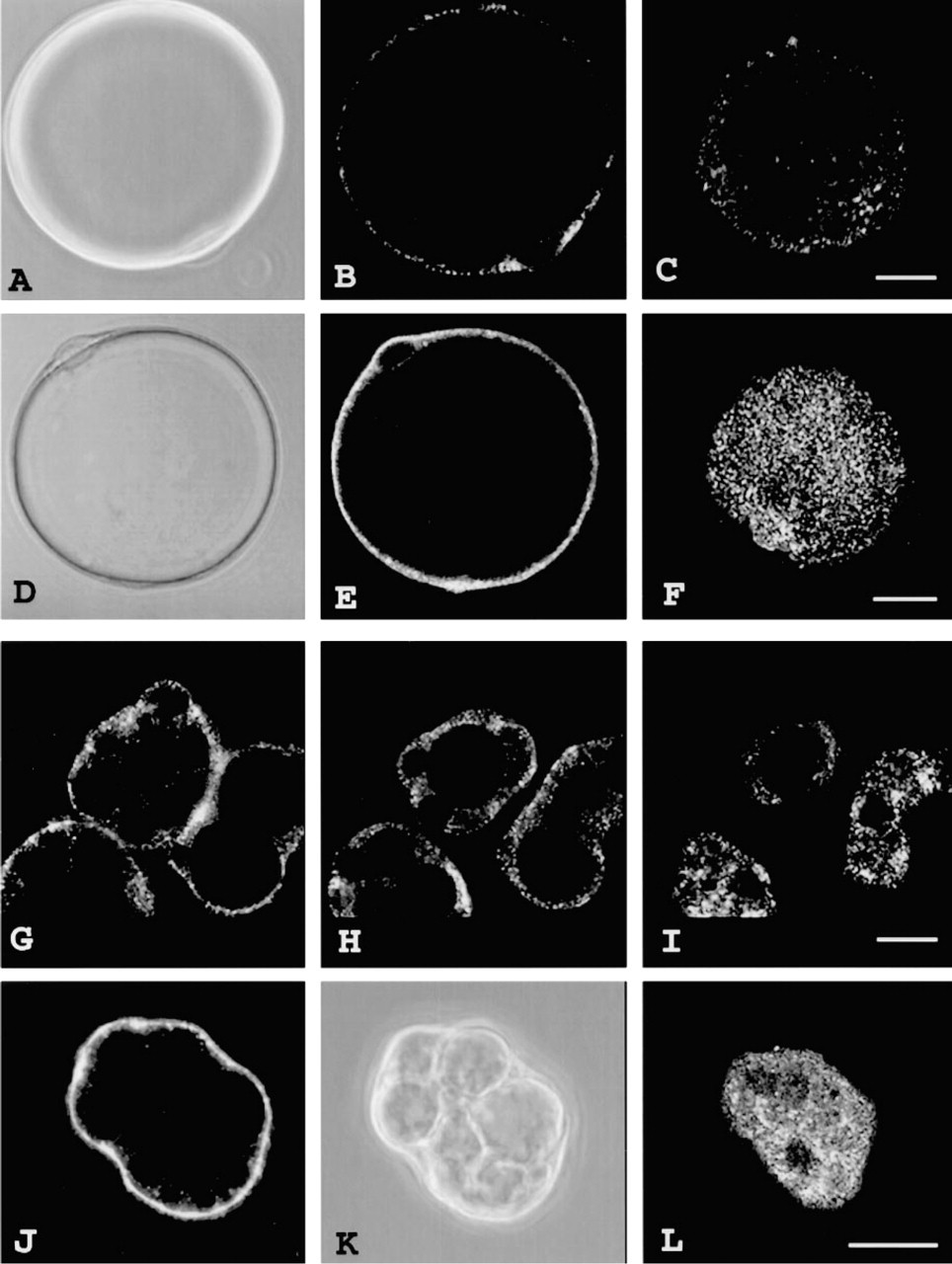
Steady-state distribution of GLUT4 in basal and insulin-stimulated isolated rat white and brown adipose cells. White (
Under phase microscopy, isolated white adipose cells appear as large round cells with a mean diameter of ∼ 80 μm and containing one large lipid droplet surrounded by a thin cytoplasmic rim (∼ 1 μm) (Figures 1A and 1D). In contrast, brown adipose cells are smaller, with a mean diameter of ∼ 40 μm; they have a central nucleus, and contain multiple lipid droplets (Figure 1K). The latter morphology greatly facilitates visualization of GLUT4 subcellular distribution, particularly the insulin-induced GLUT4 translocation, by fluorescence microscopy because of the clearer distinction between the plasma membrane and the cytoplasm.
Paraformaldehyde-fixed, saponin-permeabilized cells that are labeled with anti-GLUT4 antibodies show a characteristic staining. Optical sections (0.2 μm thick) collected in the middle of the cell and closer to the cell surface are presented for comparison of cells incubated in the absence or presence of insulin (Figure 1). Under basal conditions in both white and brown adipose cells, GLUT4 immunofluorescence is almost entirely intracellular and is found in the perinuclear region and in fine punctate spots distributed throughout the cytoplasm (Figures 1B, 1G, and 1H). Only weak staining is observed at the cell surface (Figures 1C and 1I). Both the polyclonal and monoclonal anti-GLUT4 antibodies exhibit similar patterns of immunofluorescence and efficiencies of detection. When the immunolabeling is performed using the same dilution of a particular antibody, the intensity of the signal is higher in brown adipose cells compared to white adipose cells, which suggests that the level of immunoreactive GLUT4 is higher in these cells. This is consistent with biochemical evidence that the former express higher levels of GLUT4 than the latter (Omatsu–Kanbe et al. 1996). After a 30-min treatment with insulin, a redistribution of GLUT4 immunofluorescence to the plasma membrane is observed (Figures 1E, 1F, 1J, and 1L). The marked increase in cell-surface labeling of GLUT4 is accompanied by a decrease in the punctate intracellular labeling, particularly in the perinuclear area.
To evaluate GLUT4 distribution at the cell surface, the plasma membrane was identified by the rhodamine-conjugated lectin LCA in conjunction with anti-GLUT4 labeling. In basal cells, GLUT4 staining is mainly intracellular, beneath and distinct from the plasma membrane (Figure 2A). In contrast, in insulin-treated cells, GLUT4 immunofluorescence co-localizes with that of the lectin at the periphery of the cell (Figure 2B).
Three-dimensional images of white and brown adipose cells show a heterogeneous distribution of the GLUT4 staining. Patches of very bright fluorescence are observed in the basal state (Figures 3A and 3C), and heterogeneous staining at the cell surface with areas of bright and weak immunofluorescence is seen in the insulin-stimulated state (Figures 3B and 3D).
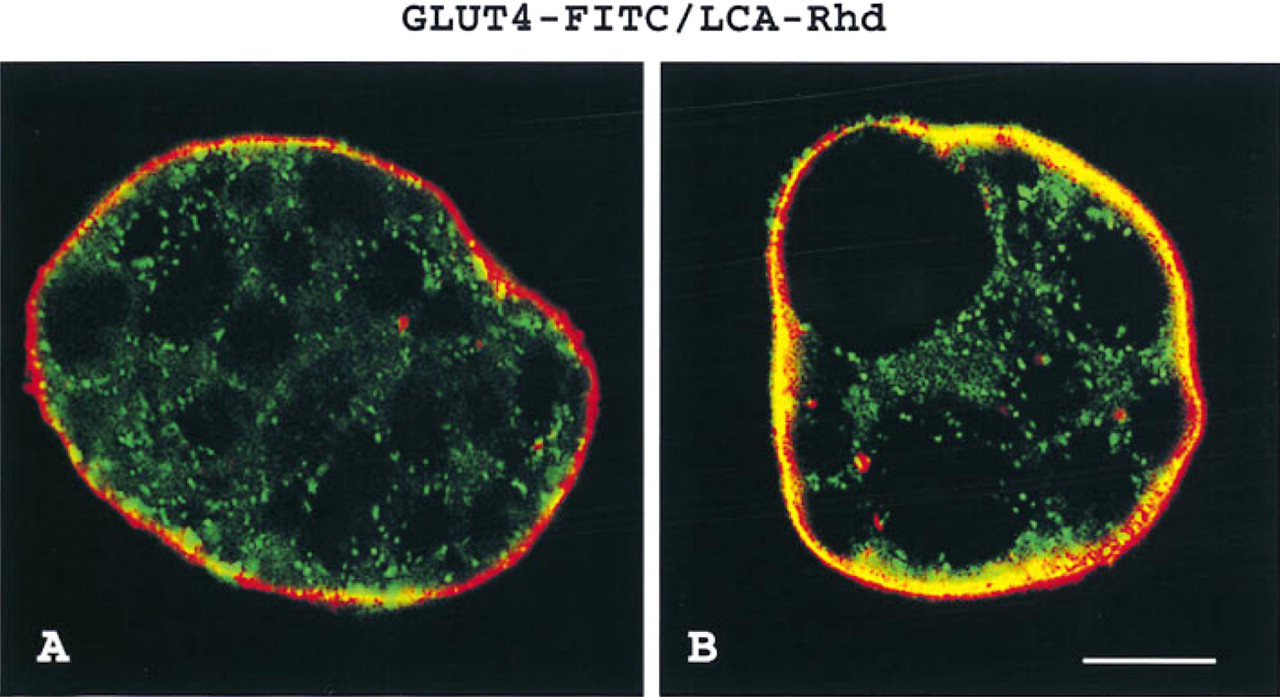
GLUT4 translocation to the cell surface in insulin-stimulated isolated rat brown adipose cells. (
The relationships between GLUT4 staining and those of various intracellular markers in the basal state, particularly for endocytic/exocytic vesicles, early and late endosomes, Golgi, and lysosomes, were examined by co-localization studies in white and brown adipose cells (Figures 4 and 5). In double labeling experiments performed in basal cells, the subcellular distribution of GLUT4 overlaps highly with that of VAMP2 and occasionally with those of clathrin and TfR but is distinct from that of lgp-120. A small fraction of GLUT4 is observed to overlap with M6PR, TGN38-mannosidase ll, and γ-adaptin staining. All the findings just described are the result of extensive analyses of many optical sections through the cells. However, we chose to illustrate images demonstrating the overlap even when these images represented a small proportion of all the focal planes examined.
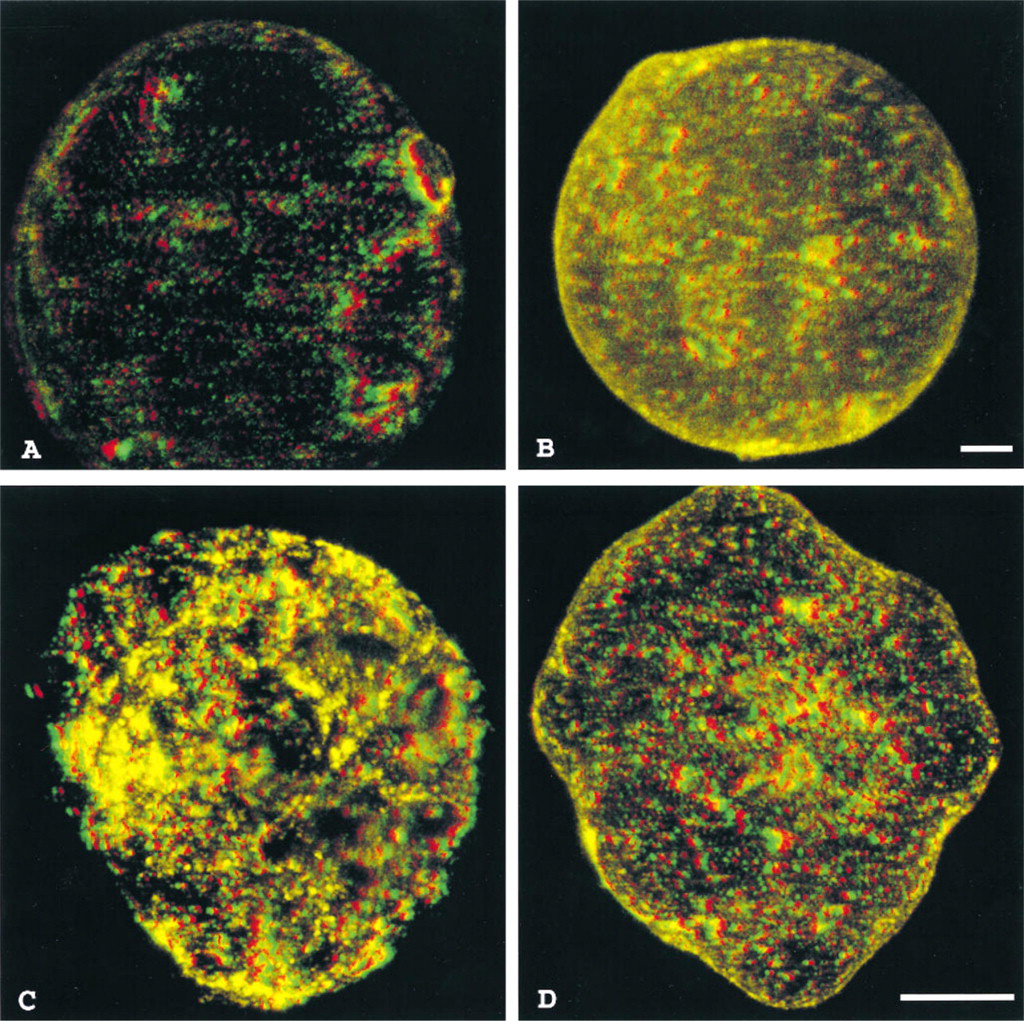
Reconstructed 3D images of whole-cell GLUT4 staining in basal and insulin-stimulated isolated rat white and brown adipose cells. Use red and green glasses (red–left; green–right) to view this image. GLUT4 labeling can be seen throughout the depth of the basal white (
Thus, for VAMP2 and GLUT4, images similar to those illustrated in Figure 4C were observed throughout most of the focal planes examined. In contrast, the overlaps for M6PR, TGN38-mannosidase ll, γ-adaptin and GLUT4 illustrated in Figures 4L, 5F, and 5I, respectively, were restricted to one or two focal steps.
In rat adipose cells, VAMP2 immunofluorescence appears in discrete punctate spots localized throughout the cytoplasm (Figure 4A). Analysis of whole cell staining reveals that GLUT4 and VAMP2 are co-localized in many vesicle-like structures (Figure 4C). A semiquantitative estimation of the degree of the overlap reveals that approximatively 50% of the total GLUT4-containing vesicles present in a cell contain VAMP2 as well. Although clathrin immunofluorescence is dispersed in fine punctate spots similar to GLUT4, a detailed analysis reveals their almost distinct immunolocalizations (Figures 4D–4D). In both white and brown adipose cells, TfR exhibit a characteristic speckled pattern that does not coincide, for the most part, with GLUT4 (Figures 4G–4G). Lysosomal membrane protein lgp-120 immunostaining is also widely distributed throughout the cytoplasm, but does not overlap at all with GLUT4 immunofluorescence (Figures 4M–4M). Although M6PR and GLUT4 display different punctate distributions, some co-localization is observed in “large” structures in the perinuclear region, presumably indicative of endosome or Golgi-type staining (Figures 4J–4J). The images illustrated here are representative of the limited overlap between GLUT4 and M6PR observed in one or two optical sections throughout the cell.
In white adipose cells, we noted a particular distribution of TGN38-mannosidase ll staining which is located discontinuously around the nucleus as well as in large spots spread towards the cell periphery (Figures 5A and 5D). GLUT4 immunofluorescence exhibits partial overlap with TGN38-mannosidase ll-positive structures (Figures 5B, 5C, 5E, and 5F). γ-Adaptin, a subunit of the clathrin adaptor AP1 at the TGN (Pearse and Robinson 1990), is also found in white adipose cells, primarily in the perinuclear region and in punctate labeling towards the cell periphery (Figures 5G and 5J). Closer examination reveals some overlap with GLUT4 in the perinuclear area (Figure 5I). In the peripheral structures, corresponding presumably to an endosomal subcompartment (Simpson et al. 1996), γ-adaptin and GLUT4 staining for the most part do not coincide (Figure 5L).
In double labeling experiments not shown, insulin treatment of white and brown adipose cells produces an increased localization of TfR and M6PR at the cell surface. Unexpectedly, VAMP2 immunofluorescence does not appear to increase at the cell surface in response to insulin despite an approximately two-fold increase over the basal level as observed in plasma membrane fractions by Western blotting (Timmers et al. 1996). In addition, the partial co-localization of GLUT4 with TGN38-mannosidase ll does not appear to change in response to insulin. However, the overlap of GLUT4 with γ-adaptin-positive structures is increased in the perinuclear region, whereas GLUT4 and γ-adaptin remain mostly distinct at the periphery.
Several control experiments were performed to confirm the specificity of these results (not shown). Staining of the cells with normal nonimmune rabbit or mouse serum instead of the primary antibodies was not detectable. Although examination of whole adipose cells allows a comprehensive evaluation of cellular distribution of immunostaining, it has the drawback of requiring permeabilization. To rule out permeabilization-related artifacts, we also stained semithin (1-μm) cryosections obtained from basal and insulin-treated cells. Treatment of sections with saponin resulted in no detectable loss of signal for GLUT4. Furthermore, these experiments confirmed the labeling pattern obtained with saponin-permeabilized cells. Another concern raised by using sequential double labeling in “intact” cells is the question of equal accessibility to different antigens that are positioned close to each other (Griffiths 1993). When antigens are closely spaced, the binding of one antibody can sterically hinder the binding of a subsequent antibody. We attempted to overcome this limitation by immunolocalizing several different marker proteins in both single and double labeling. The intensity of the staining obtained by reversing the sequence of primary antibodies was comparable in double labeling experiments.
Discussion
In the present study we have investigated by immunocytochemistry not only the whole-cell distribution of GLUT4 but also the characteristics of the membrane compartment in which GLUT4 is sequestered in the absence of insulin. Our aim was to extend our knowledge of GLUT4 trafficking pathways based on previous biochemical studies by using a complementary morphological approach. A complex study was undertaken to understand the in situ localization of GLUT4 relative to endosomal recycling receptors, Golgi, TGN, and vesicle targeting proteins. The compartmentalization and trafficking of membrane proteins have been extensively investigated and well-characterized in other cells, but we are just now starting to have the tools to perform similar studies in rat adipose cells. This approach allows us for the first time to see in situ the localization of compartments and to trace the changes in response to different stimuli. Furthermore, it overcomes some of the technical difficulties (particularly the sectioning) in studying the rat adipose cell, and it opens a new, more accessible way to investigate the subcellular GLUT4 trafficking pathways in an insulin target cell of physiological significance.
By double immunofluorescence using confocal microscopy, we have been able to analyze simultaneously the distribution of GLUT4 and various cell compartment markers. As documented here, this technique reveals with high resolution the three-dimensional distribution of whole-cell immunostaining. It thus allows definition of the precise locations of GLUT4 and cell compartment markers in areas of particular interest and extends the use of immunofluorescence from the previously described two dimensional plasma membrane lawns (Robinson et al. 1992) to the entire cell volume.
The results show an almost exclusively intracellular location of GLUT4 in basal adipose cells, with a heterogeneous patchy distribution that is more intense in the perinuclear region. The weak GLUT4 staining observed at the cell surface is clearly distinct from the lectin-labeled plasma membrane, indicating the low amount of GLUT4 localized at the cell surface under this condition. These results are consistent with previous reports using high resolution immunogold techniques (Slot et al. 1991; Smith et al. 1991) and thus validate the use of this approach to study GLUT4 subcellular traffic. The present immunocytochemical findings are associated with typically low rates of glucose transport activity as assessed by 3-O-methylglucose transport and cell-surface GLUT4 photolabeling, and corresponding low levels of GLUT4 in the plasma membrane fraction as assessed by Western blotting (Simpson et al. 1983; Holman et al. 1990; Satoh et al. 1993).
GLUT4 is highly co-localized with VAMP2, almost entirely distinct from the endosomal markers clathrin, TfR, and M6PR and clearly separate from the lysosomal marker lgp-120. Only a small proportion of GLUT4 appears to co-localize with M6PR in “large” structures most likely representing a Golgi or endosomelike pattern. Similarly, GLUT4 partially overlaps with both TGN38-mannosidase ll and γ-adaptin in perinuclear structures. These findings suggest that a small fraction of GLUT4 is present in TGN and/or late endosomes in basal cells. These compartments appear to be interconnected and a site of extensive trafficking between them. The TGN markers TGN38 and γ-adaptin are both found in endosomes as well as in the TGN in other cell types (Luzio et al. 1990; Luzio and Banting 1993; Simpson et al. 1996). In a recent study using immunoisolated vesicles in 3T3-L1 adipocytes (Martin et al. 1994), only ∼ 10% of the GLUT4 is co-localized with TGN38-positive vesicles. Immunofluorescence analysis of TGN38 and GLUT4 in these cells reveals markedly different staining patterns.
An analysis of whole-cell staining reveals that GLUT4 and VAMP2 are co-localized in many vesiclelike structures. However, it shows as well that fewer structures are labeled for VAMP2 than for GLUT4. Therefore, it is unclear whether the VAMP2-negative/GLUT4-positive vesicles represent a distinct subdomain of the GLUT4 compartment or an artifact caused by a lower labeling efficiency of VAMP2. In support of the immunocytochemical results, Western blotting analyses of immunoisolated GLUT4-containing vesicles clearly demonstrate that VAMP2 is a component of the membrane of these vesicles (Timmers et al. 1996). We conclude that in basal adipose cells GLUT4 resides mainly in specialized post-endosomal, VAMP2-positive vesicles, which constitute the insulin-responsive storage pool of GLUT4. Although the morphological identity of such a compartment in these cells is questionable, most recent reports propose that GLUT4-containing vesicles constitute a new morphological entity (Herman et al. 1994; Thorens and Roth 1996). On the basis of data obtained by overexpression of GLUT4 in insulinoma cells, these vesicles appear to be distinct from secretory or synaptic-like vesicles (Thorens and Roth 1996). Moreover, they may belong to a biosynthetic post-Golgi-regulated secretory pathway (Chavez et al. 1996).
Further support for the existence of an unique intracellular GLUT4 compartment in insulin-responsive cells comes from a study using endosome ablation and immuno-EM analysis of isolated vesicles published just after completion of our manuscript (Martin et al. 1996). These authors report that a distinct GLUT4-containing compartment is also observed in the 3T3-L1 cell line. In agreement with our results, they demonstrate that in 3T3-L1 adipocytes, under conditions in which all the recycling TfR-containing endosomes are removed, much of the GLUT4 (60%) and most of the VAMP2 (90%) are recovered in a post-endocytic compartment. With regard to characterization of this compartment, it appears clear from both studies that, in the absence of insulin, GLUT4 is segregated from the compartment containing the recycling receptors. In addition, both studies find that GLUT4 is co-localized with VAMP2.
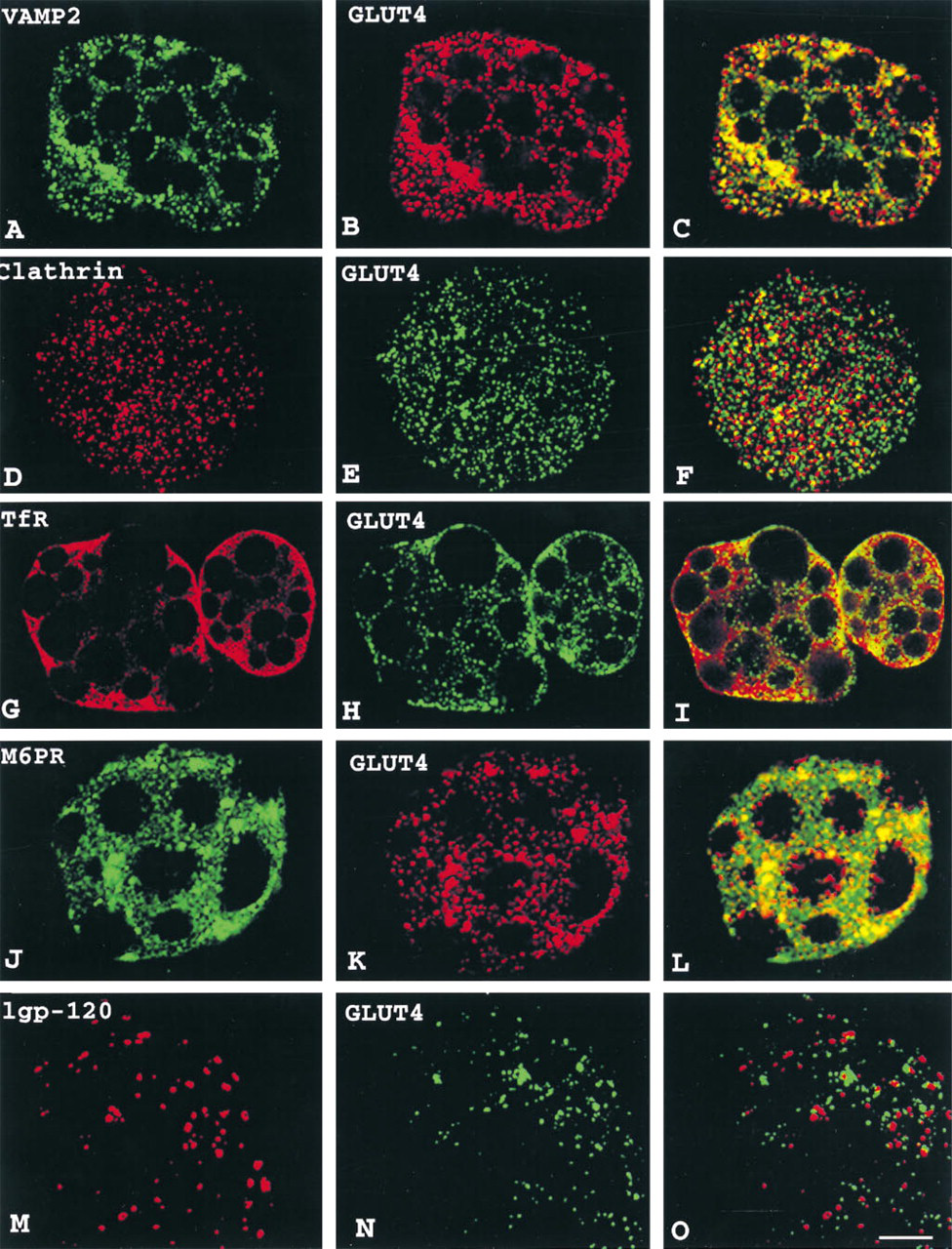
Double labeling immunofluorescence analysis of basal isolated rat white (
In addition to these similarities between the two studies, however, two differences are also particularly noteworthy. First, Martin et al. (1996) report that even in the absence of insulin a substantial proportion (∼ 40%) of GLUT4 is present in the endosomal system; our data demonstrate that the majority of GLUT4 is distinct from the TfR-containing compartment. Our results are consistent with the previous study in brown adipose tissue using quantitative immunocytochemistry at the electron microscopic level in which only 4–9% of GLUT4 is found in the endosomal system, defined by morphological criteria, in the absence of insulin (Slot et al. 1991). Most importantly, these findings may reflect essential differences in the GLUT4-containing compartment between primary cells and cells in culture (Malide et al. 1996). In rat adipose cells cultured for 24 hr, GLUT4 staining appears concentrated in perinuclear strings and large spots instead of the widely distributed punctate pattern seen in freshly isolated cells. In addition, TfR and TGN38-mannosidase ll also exhibit concentrated juxtanuclear staining and show co-localization with GLUT4 to a greater extent than observed in freshly isolated cells. We believe that in freshly isolated rat adipose cells the exclusion of GLUT4 from the early endosomal system is the result of the very efficient sorting of GLUT4 from the recycling receptors, and that this unique feature is lost when cells are cultured. One of the difficulties in studying the regulation of glucose transport and GLUT4 translocation is the lack of favorable experimental systems. Only adipose and muscle cells express endogenous GLUT4 and are the physiological insulin-target cells. In these cells, insulin leads to a rapid increase in glucose transport that is mediated by the translocation of GLUT4 to the plasma membrane.
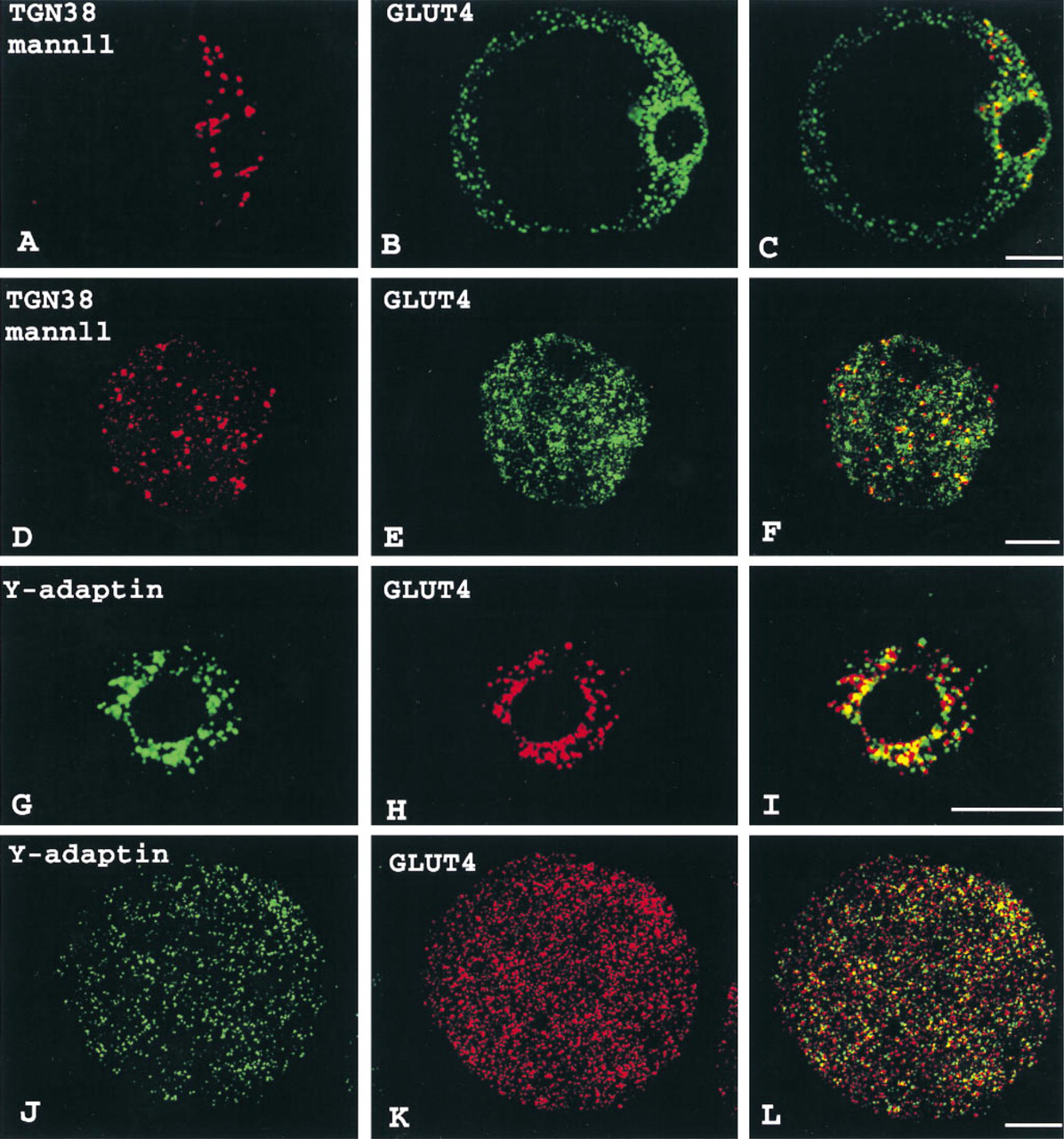
Distributions of GLUT4, TGN38-mannosidase ll, and γ-adaptin in basal isolated rat white adipose cells. Cells were incubated with a rabbit polyclonal antibody (green in
A second discrepancy between the two studies is related to the proportion of GLUT4 that is co-localized with VAMP2. In the study by Martin et al. (1996), only a quarter of the GLUT4-positive vesicles contain VAMP2 and almost half of the GLUT4-positive vesicles contain the endosomal proteins cellubrevin or M6PR. In contrast, our results in rat adipose cells show that, in the absence of insulin, approximately half of GLUT4 and VAMP2 partially share the same vesicle population. It is possible that these differences are due to the experimental approaches used: vesicles obtained in vitro by cell homogenization and examined in vitro (Martin et al. 1996) or studied in situ within the intact cells (this study).
Finally, it is interesting to note along this latter line that marked differences are observed in the immunofluorescence patterns of GLUT4 and of the “compartment markers” TfR, TGN38-mannosidase ll, and M6PR between rat adipose cells (this study) and 3T3-L1 adipocytes (Martin et al. 1994). Such differences raise several questions: (a) whether the respective compartments in these cell types are morphologically distinct; (b) how differences in morphology might affect the subcellular trafficking pathways in these cells; and (c) to what extent results obtained in a cultured cell are representative of the regulation of glucose transport in adipose cells in vivo.
Insulin induces a marked increase in the GLUT4 staining localized to the cell surface and this accounts for the 20–40-fold increase in glucose transport activity that is typically observed in both white and brown adipose cells (Vinten et al. 1976; Karnieli et al. 1981; Simpson et al. 1983; Omatsu–Kanbe et al. 1996). With maximal insulin stimulation, GLUT4 redistributes such that approximately half of the cellular GLUT4 resides in the plasma membrane (Simpson et al. 1983; Yang and Holman 1993). Interestingly, by immunofluorescence the pattern of GLUT4 staining at the cell surface appears to be heterogeneous, with patches of very bright areas suggestive of GLUT4 clusters in both white and brown adipose cells. This latter finding was not observed in previous studies using high-resolution immunogold techniques (Slot et al. 1991; Smith et al. 1991; Volstedlund et al. 1993), in which a rather random distribution of GLUT4 at the plasma membrane was described. This discrepancy may be technical in origin. Amplification due to the multiple binding of secondary antibodies may be the cause of such clusters in immunofluorescence. However, although the presence of GLUT4 in clusters remains to be clarified, these clusters could correspond either to caveolae, which are extremely abundant in adipose cells (Anderson et al. 1992) or, less likely, to hypothetical “occluded vesicles,” a primed/docked pool of vesicles adjacent to the cell surface (Vannucci et al. 1992; Satoh et al. 1993). Clusters on the cell surface itself would be consistent with the known rapid rates at which GLUT4-containing vesicles enter and leave the plasma membrane during recycling.
In insulin-stimulated adipose cells we also observe an increase in the staining of TfR and M6PR associated with the cell surface, consistent with previous studies indicating that insulin induces their translocations as well (Davis et al. 1986; Tanner and Lienhard 1987, 1989). Recent biochemical data show that 10–15% of total M6PR in white adipose cells is co-localized to GLUT4-positive vesicles, and it has been postulated that this association accounts for the insulin-induced movement of M6PR to the plasma membrane (Kandror and Pilch 1996). Nevertheless, although we find limited co-localization of GLUT4 and M6PR, it is present in the late endosomal compartment rather than in the GLUT4-containing vesicles. Similar data are obtained in 3T3-L1 adipocytes (Martin et al. 1994). In addition, previous studies from our laboratory demonstrate different kinetic rates for M6PR translocation compared to GLUT4, consistent with the possibility that M6PR and GLUT4 at least in part traffic in distinct vesicles (Appell et al. 1988).
It is tempting to propose a role for VAMP2 in targeting and regulating the traffic of GLUT4 vesicles in response to insulin. Therefore, the fact that by immunofluorescence VAMP2 staining does not display a plasma membrane pattern in response to insulin is intriguing. One plausible interpretation of this finding is that the relatively small change (twofold) in plasma membrane VAMP2 observed in our laboratory by subcellular fractionation and Western blotting is below the threshold of detectability by immunocytochemistry. Alternatively, this may be due to different rates of endocytosis in insulin-stimulated states. Assuming that the rates of exocytosis are similar, VAMP2 may be internalized faster than GLUT4, such that different steady state distributions are established.
In adipose cells incubated with insulin, partial co-localization between GLUT4 and clathrin is observed in both peripheral and perinuclear regions. These results are consistent with the idea that GLUT4 is internalized in the presence of insulin only in part by clathrin-coated vesicles and that other (un)coated vesicles also participate in this process (McKeel and Jarett 1970).
The partial overlap of GLUT4 with M6PR-, TGN38-mannosidase ll-, and γ-adaptin-positive structures suggests the possibility that in the recycling pathway GLUT4 may return as “far back” as the TGN or late endosomes before reaching their specialized VAMP2-positive subcompartment. This route is consistent with the model of two or more intracellular pools in series responsible for GLUT4 sequestration (Holman et al. 1994; Verhey et al. 1995). Studies are now in progress in our laboratory using this immunocytochemical approach to investigate insulin-modulated steps along the GLUT4 trafficking pathway.
Footnotes
Acknowledgements
We thank Mary Jane Zarnowski, Steven R. Richards, and Dr Hailan Tang for technical assistance. We are grateful to Dr Thorkil Ploug, Dr Konstantin V. Kandror, and Dr Gwyn W. Gould for making manuscripts available before publication. We thank Dr Jennifer Lippincott–Schwartz, Dr Harish Radhakrishna, and Dr Mariko Omatsu–Kanbe for many helpful discussions, and Dr Ian A. Simpson and Dr Evelyn Ralston for continuous help and encouragement and for critically reading the manuscript.