
Abstract
We describe a novel fluorescent method for the detection of receptors for chimeric proteins in tissue sections. The technique was developed using a recombinant human insulin-like growth factor (IGF-1) chimera, bearing six additional histidine residues at the carboxy-terminal end (IGF-1-His). We demonstrated that dehydration of the tissue sections was detrimental for binding and that its prevention dramatically increased sensitivity. The specificity of IGF-1-His interaction was shown by gradual abolition of the fluorescent signal in the presence of increasing concentrations of IGF-1. Combining immunofluorescence with in situ ligand binding, we showed that IGF-1-His binding corresponded to the IGF-1 receptor (IGFR-1) distribution in human fetal kidney. Moreover, incubation of the tissue sections with an anti-IGFR-1 blocking antibody abolished IGF-1-His binding, demonstrating that the interaction was mediated by the IGFR-1. The method was also used to localize the IGFR-1 in E18 rat embryo sagittal sections. The IGF-1-His binding pattern was observed in brain, cartilage, lung, skin, heart, diaphragm, and tongue, and paralleled the previously reported IGFR-1 distribution. We believe that this new non-isotopic in situ ligand binding method will facilitate rapid and accurate localization of receptors in tissue sections.
Keywords
Recently, methods using fluorescently labeled or biotinylated ligands were described for localization of receptors in tissue sections (Michel and Parsons 1989,1990; Reeves et al. 1994; Göritz et al. 1996; Ray and Ariano 1998). Binding of fluorophore-conjugated ligands is directly visualized using a fluorescent microscope, whereas the biotinylated ligands are detected using a streptavidin-conjugated enzyme and a chromogenic substrate. Although these procedures provide results rapidly, their low sensitivity often limits their application. Moreover, proteins must be labeled individually, increasing the processing time and the possibility of reducing the binding affinity to their receptor (Michel and Parsons 1989; Bentham et al. 1994; Ray and Ariano 1998).
On the other hand, when a gene of interest is cloned, the corresponding protein is often expressed fused to a peptide or protein (e.g., tag) that has unique binding properties that permit its purification from a crude cell lysate. Several different tags are available for generation of recombinant fusion proteins. The most popular include glutathione S-transferase (Smith and Johnson 1988; Guan and Dixon 1991), poly-histidine (His; Hochuli 1990), and β-galactosidase (Gray et al. 1982). Fusion or chimeric recombinant proteins often conserve their biological activity and are frequently used to study ligand-receptor interaction without removing the added peptide (Klein et al. 1997; Sheridan et al. 1997).
Here we describe a rapid non-isotopic method to locate receptors of non-labeled recombinant fusion proteins in tissue sections. The proof of concept was elaborated with an IGF-1 fused to six histidine residues on the carboxy-terminal end. We chose this system because IGF-1 is a well-documented growth factor and the His tag is one of the most widely used methods of expression of recombinant proteins.
Materials and Methods
Materials
IGF-1 was from Genentech (South San Francisco, CA). The Tissue-Tek OCT compound was from Miles (Elkhart, IN) and the Superfrost Plus Gold microscope slides were from Erie Scientific (Portsmouth, NH). The avidin-biotin blocking kit, the normal horse serum, the biotinylated horse anti-mouse IgG (H+L) antibody, and the Vectashield mounting media were obtained from Vector (Burlingame, CA). The Texas Red-conjugated anti-goat IgG antibody was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). The anti-IGF-1 receptor antibody (cat. # BAF391) used to localize the IGF-1 receptor and the anti-IGF-1 receptor antibody (cat. # AF-305-NA) used to block IGF-1 binding to its receptor were from R & D Systems (Minneapolis, MN). The mouse monoclonal anti-H6 antibody, the fatty acid ultra-free bovine serum albumin (BSA) fraction V, and the complete EDTA-free protease inhibitor cocktail tablets were from Roche Molecular Biochemicals (Indianapolis, IN). The Renaissance TSA indirect amplification kit was from NEN Life Science Products (Boston, MA). The streptavidin-conjugated FITC was from DAKO (Carpinteria, CA).
Human IGF-1 (hIGF-1) was expressed in E. coli as a full-length protein with a hexa-histidine tag at the carboxy terminus. The cell lysates were subjected to chromatography on an Ni2 +-NTA agarose column (Qiagen; Valencia, CA). IGF-1-His was eluted with a 0–500-mM imidazole gradient. Fractions containing the eluted IGF-1-His were pooled, dialyzed, aliquotted, and kept at −80C until used. The biological activity of the IGF-1-His chimeric protein was identical to that of the native IGF-1, as evaluated by its potential to induce the phosphorylation of its receptor in a KIRA assay (Sadick et al. 1999).
Tissue Preparation
The tissues were excised, rinsed with cold PBS (4C), embedded in OCT compound, and flash-frozen in liquid nitrogen-cooled (-60C) isopentane. The blocks were stored at −80C in sealed plastic bags. Before sectioning, the blocks were equilibrated at −15C to −19C for 1 hr. The 10-μm sections were cut using a CM-300 cryostat (Leica Microsystems; Deerfield, IL) and applied to the Superfrost Plus Gold microscope slides. The slides were placed horizontally on a tabletop at room temperature (RT) for 30 sec and stored immediately at −20C for a minimum of 3 days. The sections were used within 2 weeks.
In Situ Ligand Binding
Slide-mounted tissue sections were brought to RT and immediately incubated for 4 min in 35 mM acetic acid (pH 3.5) containing 3 mM CaCl2, 3 mM MgSO4, 5 mM KCl, and 1 M NaCl. The slides were then washed in HBS-C (25 mM Hepes, pH 7.2, 150 NaCl, 3 mM CaCl2, 3 mM MgSO4, 5 mM KCl, complete protease inhibitors cocktail) containing 32 mM sucrose, and the nonspecific binding sites were blocked for 20 min in HBS-C containing 3% BSA and 32 mM sucrose. The binding sites for avidin and biotin were blocked using the avidin-biotin blocking kit from Vector, and the endogenous histidine-rich sites were blocked by incubating the sections for 10 min in 1 mM NiCl. The slides were incubated in HBS-C buffer containing 3% BSA and 5 nM IGF-1-His for 1 hr and then washed three times for 1 min each with cold (4C) HBS-C buffer containing 1% BSA. The sections were fixed in PBS containing 4% paraformaldehyde for 10 min and washed with HBS-C containing 1% BSA. The endogenous antibody binding sites were blocked with 1.5% normal horse serum in HBS-C for 20 min. The slides were then incubated with 1 μg/ml anti-H6 antibody in HBS-C/3% BSA for 1 hr. Alternatively, the ligand and the anti-H6 antibody were first incubated for an hour before the mixture was added to the tissue sections. The sections were washed (three times for 1 min) with HBS-C/1% BSA and incubated for 30 min with the secondary antibody (biotinylated horse anti-mouse IgG) diluted 1:200 in HBS-C containing 3% BSA. The sections were washed three times for 4 min and fixed in PBS containing 4% paraformaldehyde for 10 min. The sections were washed with HBS-C/1% BSA and incubated for 30 min with streptavidin conjugated to horseradish peroxidase. The slides were washed three times for 1 min in HBS-C/1% BSA and incubated for 10 min with biotin-conjugated tyramide in NEN dilution buffer. The reaction was stopped by three washes of 4 min in TBS/0.1% BSA. The slides were incubated with streptavidin-conjugated FITC in TBS/0.1% BSA for 30 min and washed three times for 1 min each in TBS containing 0.05% Tween-20. The glass slides were mounted using Vectashield mounting medium.
Immunolocalization of IGFR-1
After the in situ ligand binding procedure, the sections were incubated for 1 hr with 10 μg/ml anti-human IGF-1 receptor antibody (R & D Systems; cat. # BAF391). The slides were washed three times with PBS and further incubated for 30 min with the Texas Red-conjugated anti-goat IgG antibody. The sections were washed three times with PBS and mounted using Vectashield mounting medium.
Microscopy and Imaging
The tissue sections were examined and photographed using either an Optishot or an Eclipse 800 microscope from Nikon (Melville, NY). Images were acquired using a CCD-cooled image point camera from Photometrics (Tucson, AZ). Digital image total gray value was measured using the Metamorph software from Universal Imaging (West Chester, PA).
Results
Tissue Section Processing
Fresh frozen sections of human fetal kidney were brought to RT and left to dehydrate for different lengths of time. The in situ ligand binding procedure was executed and the resulting fluorescence intensity was estimated by measuring the total gray level of the acquired digital images. As shown in Figure 1, binding of (5 nM) IGF-1-His to the cortical glomeruli rapidly decreased with increasing drying time. The highest signal was detected when the procedure was started immediately after the tissue sections were brought to RT (Figure 1A). After 2 min of drying, the signal was lowered by 52% (Figures 1B and 1E) and, after longer times, only 10% of residual fluorescence was visualized (Figures 1C-1E). Considering these results, the procedure was subsequently initiated immediately after the sections were brought to RT.
Competition of IGF-1-His Binding to Human Fetal Kidney Sections
To determine the specificity of IGF-1-His binding to human fetal kidney sections, the in situ ligand binding procedure was executed in the presence of various concentrations of IGF-1. In the absence of added IGF-1, cortical glomeruli revealed an intense fluorescent signal (Figure 2A). The fluorescent signal was gradually decreased when increasing concentrations of IGF-1 were added (Figures 2B, 2C, and 2E). At 10 nM IGF-1, the intense fluorescent signal was abolished (Figure 2C), leaving only a faint background signal comparable to the negative control executed in absence of IGF-1-His (Figure 2D). No signal was detected when the anti-His antibody was replaced by an unrelated antibody (anti-GP 120; data not shown). Furthermore, the binding of IGF-1-His was not reduced in the presence of a 20-fold molar excess of various unrelated proteins (VEGF or NGF; data not shown).
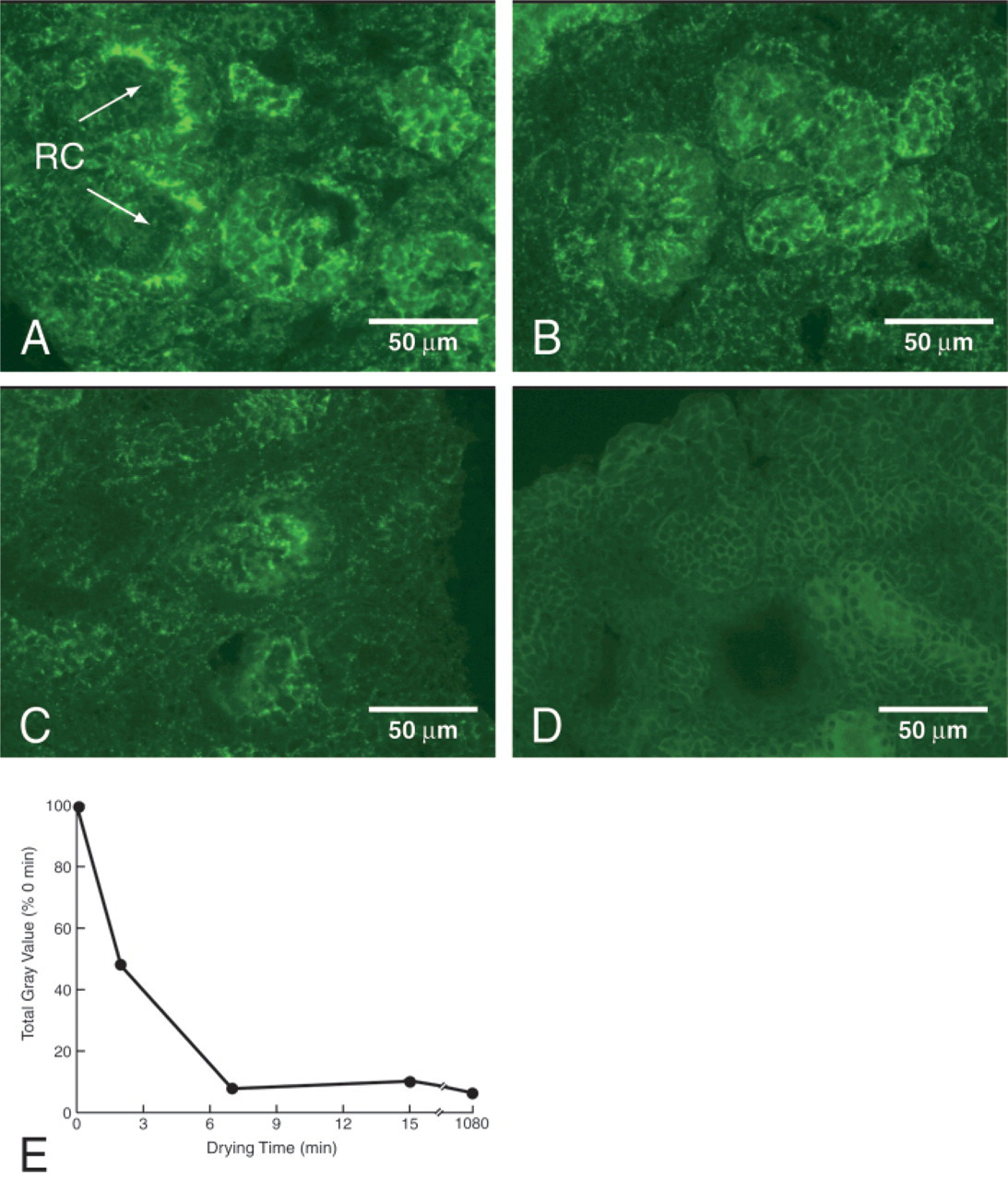
Dehydration of sections compromises IGF-1-His binding. Human fetal kidneys were sectioned as described in Materials and Methods and stored at −20C. The sections were brought to room temperature, allowed to dry for 0 min (
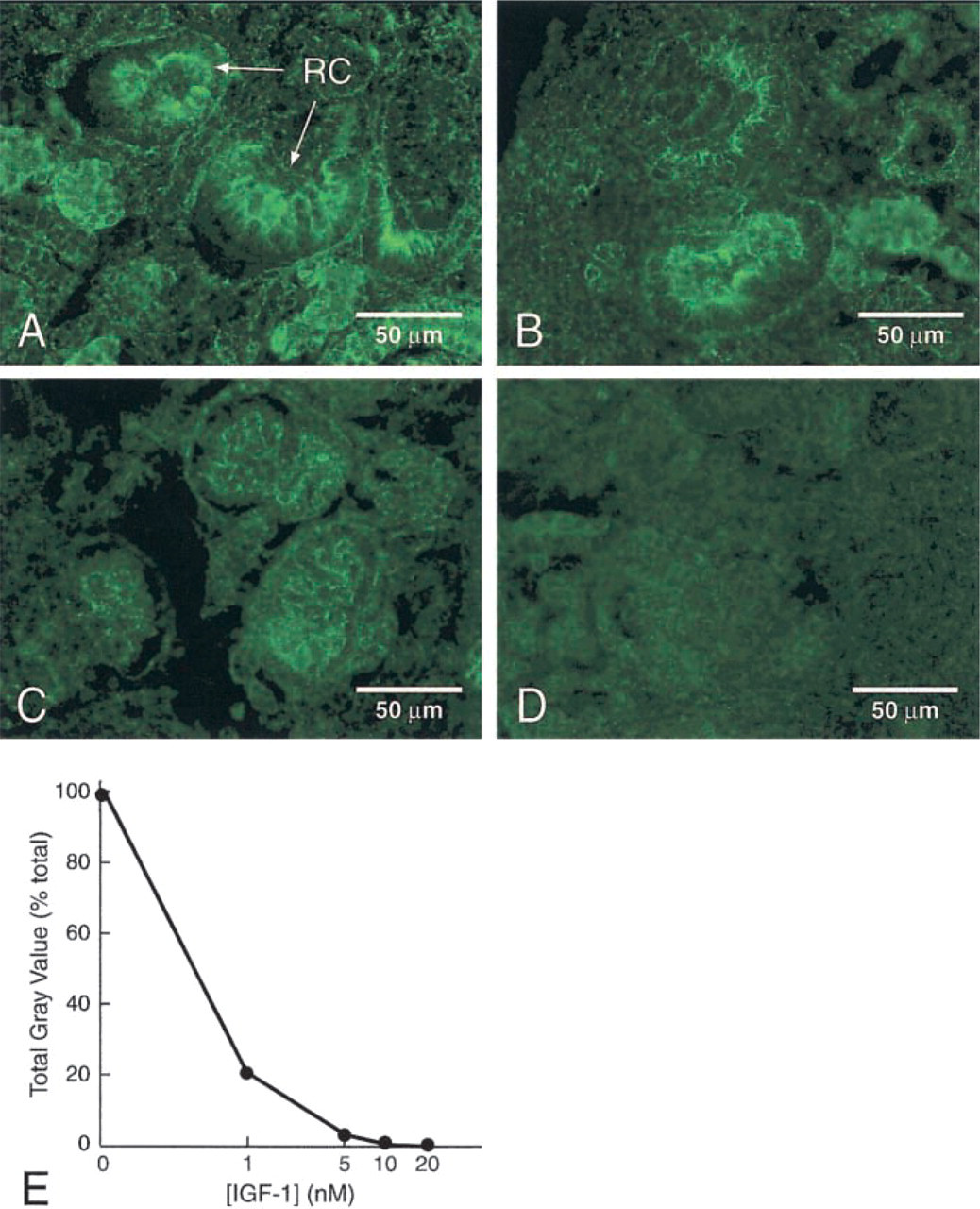
Competition of IGF-1-His binding to tissue sections. The binding of 5 nM IGF-1-His to human fetal kidney sections was assessed in the presence of various concentrations of IGF-1. (
IGF-1-His Binds the IGF-1 Receptor
To verify that the fluorescent signal corresponded to IGF-1-His binding to the IGF-1 receptor (IGFR-1), we performed immunofluorescent double staining. Immediately after the in situ ligand binding procedure with IGF-1-His, we localized IGFR-1 using a non-blocking anti-IGFR-1 antibody. As shown in Figure 3, the binding of IGF-1-His to cortical glomeruli (Figure 3A) was identical in intensity and in distribution to the immunofluorescent localization of the IGFR-1 (Figure 3B). When superimposed, the respective pictures revealed a single distribution (Figure 3C). Moreover, when the sections were incubated with a blocking anti-IGFR-1 antibody, the in situ IGF-1-His binding was abolished (Figure 3E). These results further demonstrate that the interaction of IGF-1-His is restricted to the IGFR-1. No fluorescence was observed when the addition of IGF-1-His was omitted or an unrelated primary antibody was used (Figure 3F).
Using in situ ligand binding, we localized IGFR-1 in the developing glomeruli of the renal cortex and the outer medulla and in the epithelia of the proximal convoluted tubules of human fetal kidney. In the cortical region, the fluorescence was present at the surface of the epithelial cells invaginating from the visceral layer of Bowman's capsule. The overall localization of the fluorescent signal paralleled the previously described distribution of the IGFR-1 in human kidney (Gröne et al. 1992). Taken together, these results demonstrate that IGF-1-His specifically interacts with IGFR-1 in tissue sections.
Distribution of IGFR-1 in Rat Embryo E18
The distribution of IGFR-1 was assayed by in situ IGF-1-His binding using fresh-frozen sagittal sections through embryonic day 18 (E18) rats. The fluorescent staining revealed that the IGFR-1 was widely distributed throughout the embryo. Intense, punctate fluorescent labeling was found on chondrocytes and mesenchymal cells of the cartilage primordium (Figures 4A and 4B). The lungs and the pulmonary vasculature were also stained, whereas no fluorescence was found on the epithelium of the airways (Figure 4C). A strong signal was localized in the atrial and ventricular endocardium, the trabeculi, and the myocytes (Figures 4D and 4E). The staining was seen in the endothelium and on the myocytes of the large blood vessels, the myofibrils of the tongue, the dome of the diaphragm, and the skeletal muscles (not shown). Layers II and VI of the cerebral neocortex were also stained (Figure 4F). In the epidermis, intense fluorescence was located in the germinal layer (stratum basale), whereas the outer layers revealed only weak staining (Figure 4G).
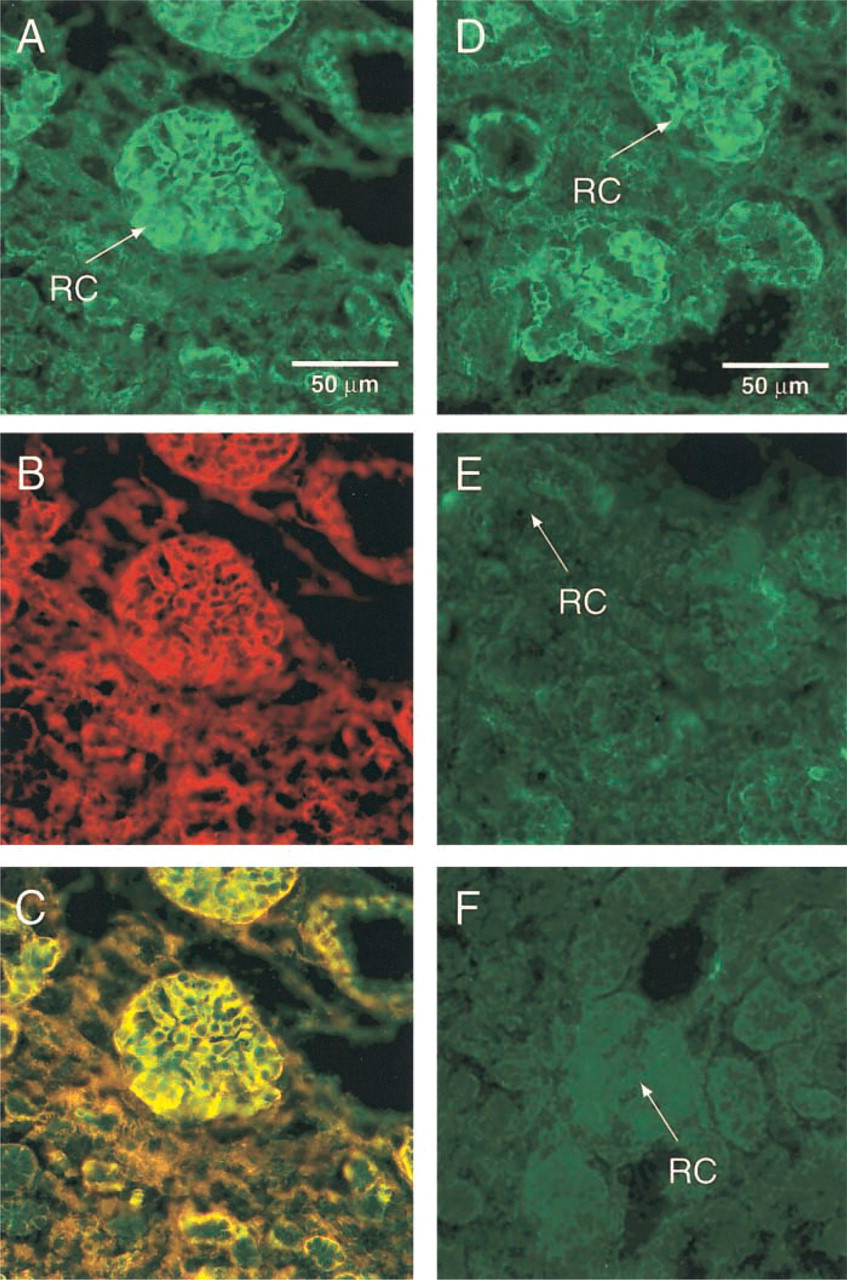
IGF-1-His binds to IGFR-1 in tissue sections. in situ IGF-1-His binding and immunofluorescent detection of IGFR-1 with a non-blocking anti-human IGFR-1 antibody were sequentially executed on human fetal kidney sections. (
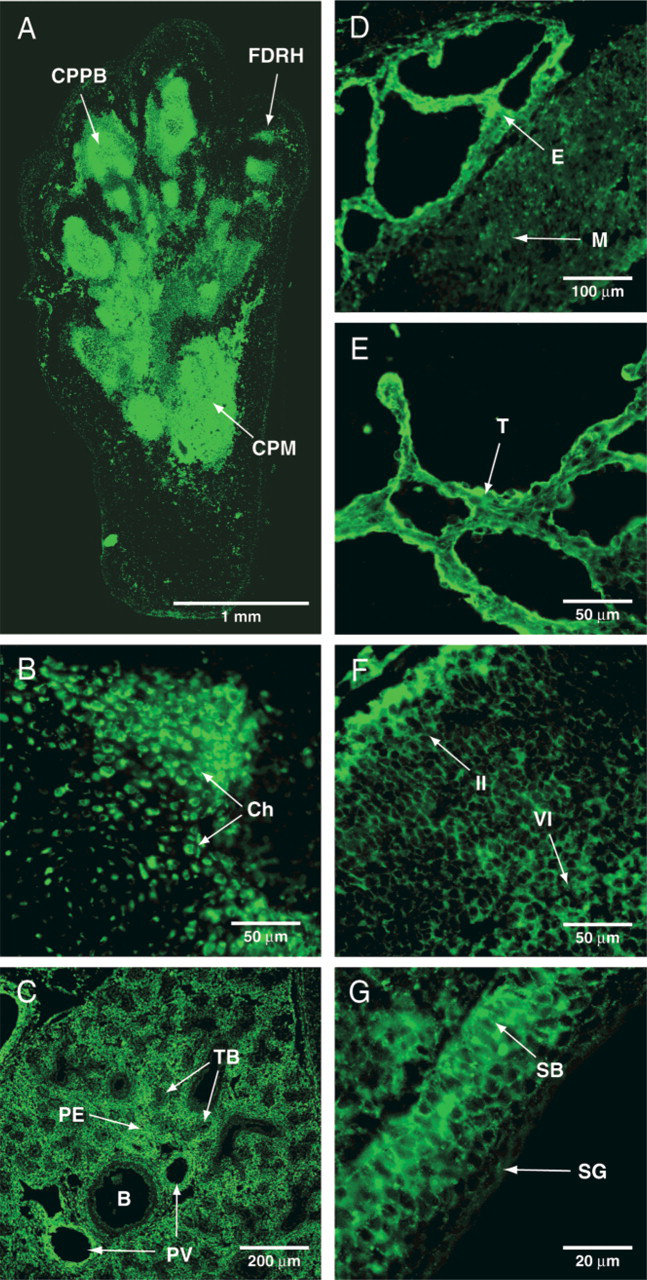
Distribution of IGFR-1 in E18 rat. The in situ ligand binding procedure was executed on sagittal sections of E18 rat as described in Materials and Methods. Strong fluorescent staining was observed in the chondrocytes and mesenchymal cells of the cartilage primordium of the hind-limb (
Discussion
We describe here a novel approach for localizing receptors of fusion proteins in tissue sections. The protocols for the preparation and the storage of the tissue sections were designed to minimize the loss of binding activity. For this reason, tissues were not fixed but were embedded in OCT immediately after excision, stored at −20C, and used within 2 weeks. Prolonged storage resulted in dehydration of the tissue, compromising IGF-1-His binding (data not shown). Similarly, the binding of IGF-1-His was greatly reduced when the sections were allowed to dry when brought to RT (Figure 1). Treatments with periodic acid-based endogenous peroxidase blocking solutions or fixation with acetone, ethanol, or 4% formaldehyde equally abolished binding (data not shown). Overall, treatments that denatured, modified, or damaged cell receptors reduced IGF-1-His binding.
Because it promotes the adhesion of tissues to microscope slides, dehydration of sections is often integrated into the receptor autoradiography protocol. Desiccation for various lengths of time, storage in the presence of desiccant, or drying of the tissue sections was reported by several authors (Millan et al. 1986; Lesniak et al. 1988; Michel and Parsons 1989; Kar et al. 1993; Winzer-Serhan et al. 1997). Although convenient, the dehydration of the tissue sections irreversibly denatures a portion of the receptors. The reduced number of endogenous binding sites artificially lowers the sensitivity of the detection method by decreasing the number of labeled molecules able to bind. In previously reported protocols (Lesniak et al. 1988,1991; Bohannon et al. 1988; Gröne et al. 1992; Kar et al. 1993; Chin et al., 1994) the use of highly sensitive radiolabeled IGF-1 compensated well for the diminished sensitivity associated with a decreased number of biologically active receptors. Although such a ligand allows the localization of receptors in dehydrated sections, the reduced binding activity imposes a longer exposure time (1–14 days). Because no binding could be detected in dehydrated sections, it is clear that the non-isotopic reporter system used in the current method has a reduced sensitivity compared to the classical receptor autoradiography. On the other hand, by avoiding the dehydration of the tissue sections and preserving the biological activity of the endogenous receptors, the sensitivity of the technique was greatly increased.
The current technique is more sensitive than several non-isotopic receptor detection methods. With some previously reported protocols, no binding could be detected in tissues with moderate levels of receptors (Reeves et al. 1994). In this case, the in situ IGF-1-His binding gave an overall staining pattern well representing the known IGFR-1 distribution. Some techniques uses concentrations of ligand between 8- and 1000-fold greater than the dissociation constant of the ligand-receptor interaction (Michel and Parsons 1988, 1989,1990; Zhang et al. 1993; Bentham et al. 1994; Ahmed et al. 1995). For near-maximal saturation of the receptors, there should be no need to use concentrations of the ligand that are more than about fivefold greater than the dissociation constant of the ligand-receptor interaction (Reeves et al. 1994). Reported values for the IGFR-1 are in the range of 0.1–1 nM (Siddle and Soos 1999). In this study, 5 nM IGF-1-His was used to ensure a near-maximal saturation of the IGFR-1 with the lowest affinity reported. Lower concentrations of ligand generated a weaker signal, whereas higher concentrations did not show additional benefits (data not shown). For optimal sensitivity of this method, the emphasis must be placed on high biological activity and near-maximal saturation of the receptors.
It is difficult to assess the minimal number of copies of the receptor required to generate a detectable signal. A bright fluorescent signal was detected when the in situ ligand binding procedure was executed on pellets of MCF-7 cells (human breast cancer cell line; data not shown). Knowing that 90,000 IGFR-1 are present at the surface of MCF-7 cells (Favoni et al. 1998), we can expect the in situ ligand binding procedure to be able to detect at least that many receptors on a cell surface.
The specificity of IGF-1-His binding was demonstrated by the gradual disappearance of staining after addition of increasing concentrations of non-tagged IGF-1 (Figure 2). Moreover, the staining remained unchanged in the presence of a 20-fold molar excess of unrelated proteins (VEGF or NGF; data not shown).
In situ IGF-1-His binding to human fetal kidney sections paralleled the distribution of the IGFR-1 showed by immunofluorescence using an anti-IGFR-1 antibody. When both detection methods were executed consecutively, the dual staining revealed a single distribution pattern (Figures 3A-3C). Moreover, IGF-1-His binding was abolished when the human fetal kidney sections were first incubated with a specific anti-IGFR-1 blocking antibody (Figures 3D-3F). The observed binding pattern conformed to the expected localization based on the previously reported identification of IGF-1 receptors in various renal cell types. The IGFR-1 was characterized in cells from renal tubules and glomeruli (Pillion et al. 1988) and in endothelial and epithelial cells from murine glomeruli (Conti et al. 1989). Moreover, the distribution of IGF-1-His binding was identical to the previously reported IGFR-1 binding of [125I]-IGF-1 in human fetal kidney (Gröne et al. 1992; Chin et al. 1994). Together, these results demonstrate that the binding of IGF-1-His in tissue sections is localized to the IGFR-1.
In the course of this study we identified a recurrent source of nonspecific fluorescent staining. Further analysis revealed that the anti-His monoclonal antibody bound directly to endogenous histidine-rich components of the vasculature. The histidine-rich glyco-protein (HRG) is composed of almost 10% histidine by weight and is present at high concentrations in platelets and serum of several species (Haupt and Heimburger 1972; Heimburger et al. 1972; Morgan 1981; Leung et al. 1983). It is very likely that the rat orthologue of the HRG, or a similar protein containing a high proportion of histidine residues, was recognized by the anti-His monoclonal antibody during the in situ ligand binding procedure. We could successfully use the previously demonstrated affinity of the histidine rich domain of HRG (Morgan 1981,1985) to prevent its interaction with the antibody. Incubating the tissue sections with nickel chloride before the in situ ligand binding procedure abolished the nonspecific staining of endogenous histidine-rich components.
The in situ ligand binding technique was subsequently used to localize the IGFR-1 in rat E18 embryo sagittal sections (Figure 4). An intense signal was associated with the chondrocytes and the mesenchymal cells of the cartilage primordium. This is consistent with the role of IGF-1 in bone growth (Schmid et al. 1991). Similarly, Liu et al. (1993) reported that mice carrying a null mutation in the gene encoding the IGF-1 receptor demonstrate developmental delays in ossification of the cranial and facial bones, as well as bones of the trunk and extremities. We also localized intense staining in the atrial and the ventricular endocardial myocytes, and a fainter staining in the tongue and skeletal muscles. IGF-1 was shown to stimulate myogenic differentiation (Florini et al. 1991), and mice with an inactive IGF-1 show a delay in muscle maturation and a general muscular dystrophy that is most easily seen in the diaphragm, heart, and tongue (Powell-Braxton et al. 1993). In addition, it was demonstrated that mice with a null mutation in the IGF-1 receptor gene show a reduced body weight that appears to be the consequence of a decrease in myocyte number (Liu et al. 1993). An intense fluorescent signal was also localized in the fetal lungs. The synthesis of IGF-1 by fetal lung explants was previously shown (Davenport et al. 1988). Most importantly, Powell-Braxton et al. (1993) have shown that mice with an inactive IGF-1 are dead at birth because of their inability to breathe. The cause of this inability, however, was not specified. On the other hand, Liu et al. (1993) reported that mice carrying a null mutation of the gene for IGFR-1 are born alive and, despite visible efforts to breathe, die within minutes of respiratory failure. Binding was also found in the cerebrum, with a distribution that agrees with the previously reported localization of IGFR-1 in the rat brain (Bohannon et al. 1988; Lesniak et al. 1988; Kar et al. 1993). In the epidermis, the strata basale was intensely stained, whereas the outer layers showed only weak fluorescence. This distribution correlates with the immunostaining of the IGFR-1 that was previously reported (Krane et al. 1991). The IGFR-1 located in the germinal layer of the epidermis was proposed to play an important role in epidermal cell proliferation during skin development by mediating the action of the IGF-1 produced in the dermis (Liu et al. 1993). The overall staining pattern given by the in situ IGF-1-His binding in rat embryo E18 and in human fetal kidney is in good agreement with the reported IGFR-1 distribution.
In conclusion, we demonstrated that the non-isotopic in situ ligand binding procedure described here can be used to rapidly and specifically localize unlabeled chimeric protein receptors in tissue sections.
Footnotes
Acknowledgments
We thank Dr Kenneth J. Hillan and Dr Anne M. Ryan for pathological analysis of tissue sections, and Robin E. Taylor and Barbara D. Wright for technical assistance.
This publication is dedicated to the memory of Michael J. Cronin, who initiated the in situ ligand binding project.