
Abstract
Lipocortin 1 (LC1, annexin 1) has received considerable attention as a substrate for protein kinases, as a Ca++- and phosphatidylserine-binding protein, and as a mediator of glucocorticoid anti-inflammatory effects. However, there has been confusion over localization of LC1 immunoreactivity (LC1-ir), which reportedly localizes to neurons and/or to astrocytes or microglia in rat brain. To test whether these contradictory data arise from unusual properties of the antigen, we developed a novel brain slice model to determine fixation and staining variables. The specificity of anti-LC1 sera was ensured by pre-absorption and affinity purification with immobilized recombinant LC1. Specific LC1-ir was detected in ramified microglia of brains perfused with acidified aldehydes and embedded in paraffin. However, commonly used immunohistochemical procedures have unexpected profound effects. LC1-ir was eliminated by fixation with neutral/alkaline aldehydes, by freezing before strong acid-aldehyde fixation, or by staining without partial de/rehydration before the primary serum. The sensitivity of LC1 epitopes to proton and water activities may reflect molecular properties important to LC1's roles in vivo. True LC1-ir was not detected in normal neurons or astrocytes.
Lipocortin 1 (LC1), a Ca++-binding protein with phospholipase A2 inhibitory activity, was purified by Fava and Cohen (1984) as a substrate for epidermal growth factor (EGF) receptor tyrosine kinase activity. It also received considerable attention as a putative mediator of the anti-inflammatory effects of glucocorticoids (DiRosa et al. 1985). LC1 proved to be prototypic of a family of proteins, now known as annexins, that selectively bind Ca++ and phosphatidyl serine. The biological properties of annexins are apparently regulated by tyrosine phosphorylation (Haigler et al. 1992).
Annexin research has been unusually controversial (reviewed by Flower and Rothwell 1994; Raynal and Pollard 1994). Glucocorticoid responses were apparent in some experimental systems but not others. Properties detected in vivo were not apparent in vitro, and vice versa. Inhibition of phospholipase A2, the hallmark activity of annexins, involved sequestration of the substrate rather than inhibition of the enzyme.
As a possible key to the functions of EGF in situ, we have studied immunohistochemical localization of LC1 in embryonic development and in pathophysiological responses (McKanna et al. 1992; McKanna and Cohen 1989). It became apparent that LC1 immunoreactivity (LC1-ir) is very sensitive to fixation conditions. It was not preserved by buffered aldehydes, it required strong acidic fixatives, such as Carnoy's or Bouin's, and it was labile when fixed overnight.
In other hands, the conditions we found unacceptable reportedly preserved LC1-ir in interesting loci. For example, the literature contains uncontested reports that LC1 in rat brain localizes to neurons and/or to astrocytes or microglia (Mullens et al. 1994; McKanna 1993a; Strijbos et al. 1991). These conflicting data constitute the most confusing and controversial aspects of LC1 research.
Assuming that the conflicting results were unlikely to be due to mere error, we hypothesized that they might reflect interesting and unusual properties of the antigen, as suggested from other disciplines. Biochemical studies detected Ca++-dependent and Ca++-independent binding of LC1 to membranes (Myatt et al. 1992). Biophysical studies showed that annexins exhibit proteolipid-like properties and differentially partition to organic phases under acidic but not neutral conditions (Genge et al. 1991). Because immune and inflammatory responses are suppressed by LC1 (Perretti et al. 1993; Sakata et al. 1990), raising specific anti-LC1 sera may not be trivial, i.e., the most interesting epitopes may suppress antigenicity. We concluded that several fundamental molecular properties of LC1 could influence its immunolocalization and that better understanding of these factors could illuminate the roles of LC1 in vivo.
To investigate the effects of fixatives and histological protocols on LC1 and its antigenic epitopes, we developed a novel brain slice model system and used it to test more than 200 procedural variables. For example, slices can be rapidly transferred from one solution to another, or frozen, heated, or cooled, in elaborate sequences with excellent reproducibility. A thorough series of experiments using slices demonstrated that some common immunohistochemical methods, including freezing for cryosectioning, fixation with neutral aldehydes, and dehydration of the tissue, can cause profound and unexpected changes in LC1 immunoreactivity.
Because antibody specificity was a critical factor, we used LC1 from both natural and recombinant sources for pre-absorption controls and affinity purification of our anti-LC1 sera. The results strongly indicate that LC1 in normal rat brain is restricted to and characteristic of microglia.
Materials and Methods
Tissue Preparation
For optimal preservation of LC1-ir, adult Long-Evans rats were deeply anesthetized with sodium pentobarbital and perfused transcardially with 50 ml of heparinized saline (0.9% NaCl + 0.02% NaNO2 + 500 IU heparin), followed by 250 ml of FPAS fixative delivered by a gravity head of 1–2 m over 15–20 min. FPAS (formalin-periodate-acetate-salt) contained final concentrations of 10% formalin (3.7% formaldehyde + 1.5% methanol), 10 mM sodium m-periodate, 40 mM phosphate buffer, 1% acetic acid, and 0.9% NaCl. Equivalent excellent preservation also was achieved with GPAS (2.5% glutaraldehyde + PAS as above).
Immediately after perfusion, brains were removed from the skull, cut coronally into 2–4-mm-thick blocks, and post-fixed for 6 hr. The blocks were dehydrated through a 30, 50, 70, 80, 90, 95, 100% ethanol series and remained in absolute ethanol overnight at 4C. The following day they were vacuum-embedded in paraffin.
Immunohistochemistry
Sections 10 μm thick were mounted on glass slides, deparaffinized with xylenes, treated with 0.3% H2O2 in absolute methanol for 15 min to eliminate endogenous peroxidase, rehydrated through the ethanol series, and stained for immunohistochemical localization of LC1. After saturating nonspecific IgG binding sites with 10% goat serum in PBS (30 min), primary antiserum (rabbit) diluted 1:8000 in PBS + 1% goat serum was applied (overnight at 4C in a humid chamber). After washes in PBS (three times for 10 min), the bound rabbit immunoglobulins were localized with ABC Elite Vectastain reagents (Vector Laboratories; Burlingame, CA) or LAB-SA (Zymed Laboratories; So. San Francisco, CA), using diaminobenzidine as chromogen. A light counterstain of Toluidine blue also was applied in most cases.
Cryosections
After fixation as above, brain blocks were rinsed in PBS, cryo-protected overnight with 25% sucrose in PBS, frozen, and cryosectioned at 10–20-μm thickness on a sliding microtome. In our standard protocol, staining of the sections (dried on slides and stored at −20C) began with initial dehydration through the series of ethanols to enter the protocol above at the H2O2-methanol step; i.e., before exposure to the primary antibody. This initial dehydration followed by rehydration is referred to as “DeRe.”
Brain Slices
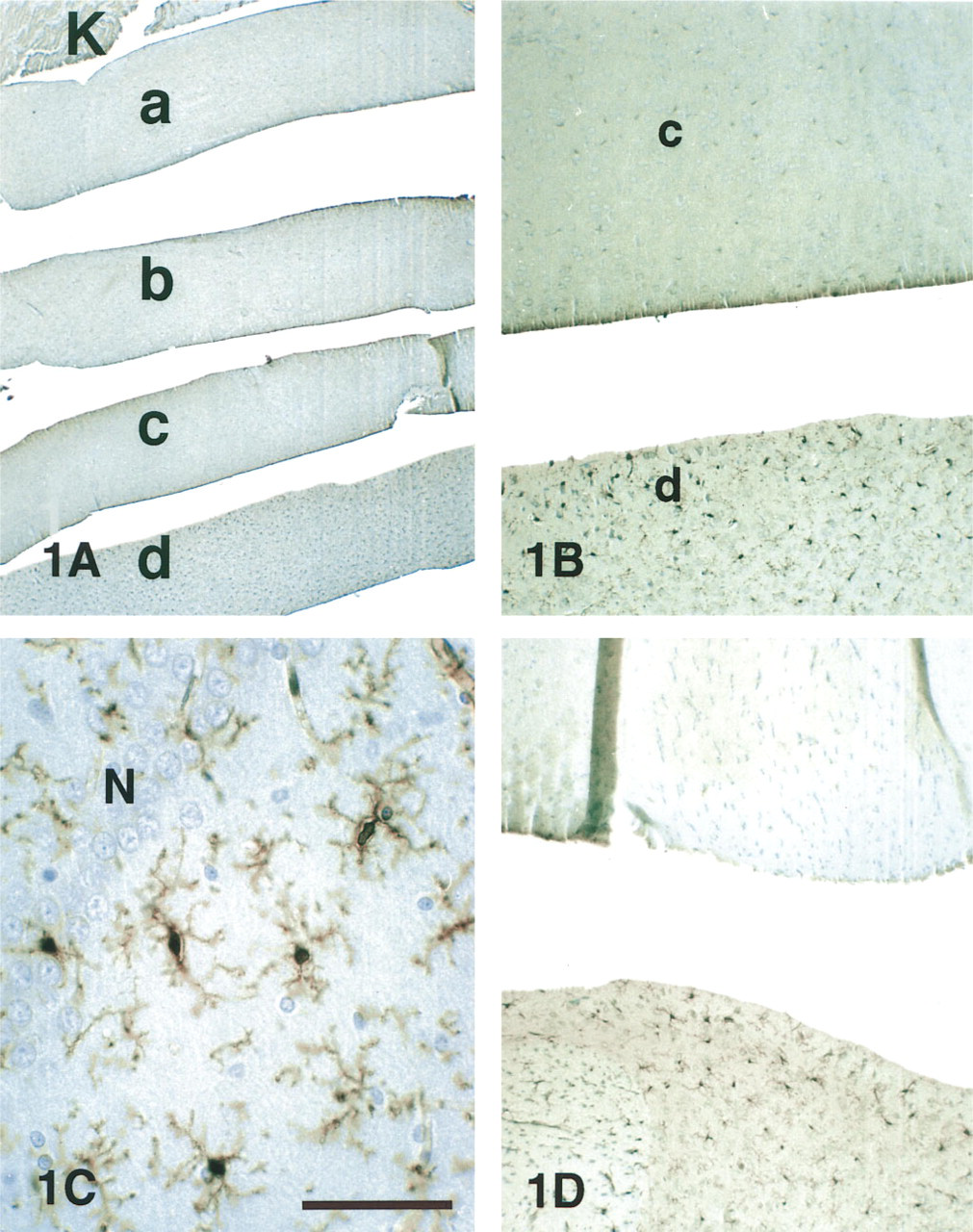
Because it would not be practical to use individual animals for the anticipated experiments with multiple fixatives and sequential methods, we tested brain slices as used for electrophysiology and pharmacology experiments (Lovinger et al. 1994) as a model system. Euthanized rats were perfused with chilled heparinized saline and the brains were removed and immersed in ice-cold PBS. Within 10 min, slices were prepared using a Vibroslice (World Precision Instruments; Sarasota, FL) and bisected to yield symmetric hemislices for experimental and control specimens. After experimentation, the slices were dehydrated and infiltrated according to the same protocols as above. However, at the point of embedding the ordered slices were placed in tandem on edge, usually propped against a larger piece of perfused kidney (Figure 1A). Sections from such blocks contained multiple specimens cut at identical thickness and subjected to identical staining procedures. The staining in the kidney served as a reference standard for quantification of LC1-ir in the brain experiments. Although many of the more than 200 variables were tested in single pilot trials, all critical aspects were repeated at least three separate times.
Antisera
The most effective anti-LC1 serum tested to date is a polyclonal rabbit serum against LC1 antigen (Ahn et al. 1988). Unlike antigen prepared by use of an EDTA eluate of Ca++-dependent LC1 bound to membrane and cytoskeleton (Fava and Cohen 1984), the Bienkowski methods (Ahn et al. 1988) treated the particulate fraction with Triton X-100. This detergent treatment presumably liberates the so-called Ca++-independent species of LC1 (Myatt et al. 1992). Little is known regarding different LC1 isoforms. However, the serum prepared by Ahn shows far more avidity for microglial LC1 than several other sera raised against Ca++-sensitive LC1. Rabbit polyclonal serum raised against recombinant LC1 provides good microglial staining and is available commercially (Zymed Laboratories).
Antibody Specificity Controls
Lipocortin for pre-absorption controls was prepared as the Ca++-sensitive eluate from placental membranes (Fava and Cohen 1984). Recombinant LC1 was purified from yeast transfected with the cDNA for human LC1 kindly provided by Dr. B.H. Chung (Korea Research Institute of Bioscience and Biotechnology) and Dr. Y. Giga-Hama (Asahi Glass Co. Research Center, Yokohama). These two sources allowed testing of rLC1 from different transfection vectors in quite different yeasts, Saccharomyces diastaticus from Dr. Chung (Tohda et al. 1994) and Schizosaccharomyces pombe from Dr. Giga-Hama (Nam et al. 1994).
For pre-absorption controls and affinity purification using the yeast as a substrate, recombinant and control yeast were fixed with GPAS, quenched with 0.1 M glycine in PBS, and frozen three times in this solution to perforate the cell walls. For each sample, 1 ml of packed yeast was incubated with 1 ml of anti-LC1 at 1:2000 dilution. After 2 hr the pre-absorbed supernatant was reserved and the yeast washed five times in PBS. Bound antibody was extracted by rapid mixing with 1 ml of 0.2 M HCl at pH 1.7 and sedimentation of the yeast. The supernatant was immediately pipetted into an equal volume of double-strength PBS, pH 7.9. The resulting solution had pH near neutrality and was used at full strength (with added 1% goat serum) for immunohistochemistry.
Quantitative Image Analysis
Density of the LC1-ir was analyzed using BIOQUANT computerized video image analysis systems (R&M Biometrics; Nashville, TN). Mean density on a scale of 0–255 was determined for >50 cells per specimen and was standardized as a percentage of the density range defined by the kidney specimen on the same slide. Because previous studies (McKanna et al. 1992) have demonstrated consistently strong LC1-ir in the medullary collecting ducts of normal rat kidney, the density of this staining was taken as 100% and assigned a rank of 4+; unstained regions of the kidney cortex defined the density of background (0% = −). Other ranks were assigned from intervals on a linear scale from these endpoints: <10%, −; 10–20%, +/-; 21–40%, +; 41–60%, 2+; 61–80%, 3+; and 81–100%, 4+.
Results
Brain Slice Model
To determine whether immersion-fixation of brain slices could serve as a model system to investigate the preservation of LC1-ir, pilot experiments varied the fixatives and the interval between slicing and fixation. FPAS or GPAS, the “best” fixatives for perfusions, preserved strong LC1-ir (4+) in an extensive population of stellate glia immersion-fixed slices immediately (Figure 1B). To determine whether the speed of transfer from slicing bath to fixative affected LC1-ir, slices were incubated at 4C in the bath or at 37C in oxygenated artificial cerebrospinal fluid (aCSF) for 50 min before fixation. LC1-ir staining intensity was equivalent in control and incubated slices. Hemislices from the same brain processed in parallel revealed the effects of some elementary fixation variables (Figure 2). Some form of fixation was necessary; no LC1-ir (-) was preserved in slices placed immediately in the ethanol dehydration series or pickled for 5 hr in PAS, the acidified buffer containing no aldehydes.
The immunostained cells in the slices were morphologically characteristic of microglia, as shown previously for tissue sections (McKanna and Fedoroff 1996). They were distinct from and approximately half as numerous as astrocytes identified by $100-β, they were dispersed (i.e., somata rarely contiguous), and they stained with other microglial markers, including GSA B4-isolectin and phosphotyrosine. In LC1-positive cells, the reaction product consistently filled the perinuclear cytoplasm and stained all the way to the tips of the ramifications (Figure 1C).
Penetration of the fixative into the slices appeared to be thorough and effective. Background immunoreactivity was very low in the median ~400 μm of the slice. However, zones at each surface of the slice (~50 μm thick) stained more densely (Figure 1B). Close examination of these superficial zones revealed many stained particles 0.5–3 μm in diameter, suggesting vesiculation of microglial processes severed during slicing.
We concluded that brain slices immersed in acidified aldehyde fixatives, embedded in paraffin, and stained with anti-LC1 serum at 1:8000 dilution provided a useful model. Although periodate was not necessary in the fixatives, we used it routinely because it appeared to improve penetration in glycosylated tissues, such as brain and embryos. Because both FPAS and GPAS provided excellent preservation of LC1-ir, their composition was varied in subsequent experiments.
Fixative Variables and Freezing: Paraffin-embedded Tissue
pH. Using the excellent preservation of LC1-ir (4+) by GPAS as control, we systematically studied the effects of pH in brain slices (Figure 2). To examine higher pH, the control fixative containing 1% acetic acid was neutralized with NaOH (1 N), essentially producing sodium acetate (~160 mM). With neutral fixative (pH 7.0–7.2), LC1-ir was barely detectable (+/-) (Figure 1B, top). At slightly alkaline levels (above pH 7.5), LC1-ir disappeared (-) (Figure 1D, top). Identical results were obtained with fixatives buffered to pH 7.2 and 7.6 with 0.1 M phosphate.
To test whether the influence of pH on LC1-ir preservation depended strictly on proton activity (H+) or might also be affected by organic anions, a gallery of acids was investigated. At concentrations of 1–5%, formic, acetic, n-butyric, i-butyric, oxalic, cacodylic, tannic, and trichloroacetic acids at pH 3–5 were very effective in preserving LC1-ir (3+ or better). Even a simple concoction of 10% formalin combined with 0.9% NaCl and brought to pH 4 with HCl preserved LC1-ir well (3+). Although these experiments did not rule out the complex tissue anions generated when fixatives interact with brain, no requirement or preference for specific buffer anions was detected in the preservation of LC1-ir. With aldehyde fixatives, only proton levels appear to be important.
Slice preparations. (
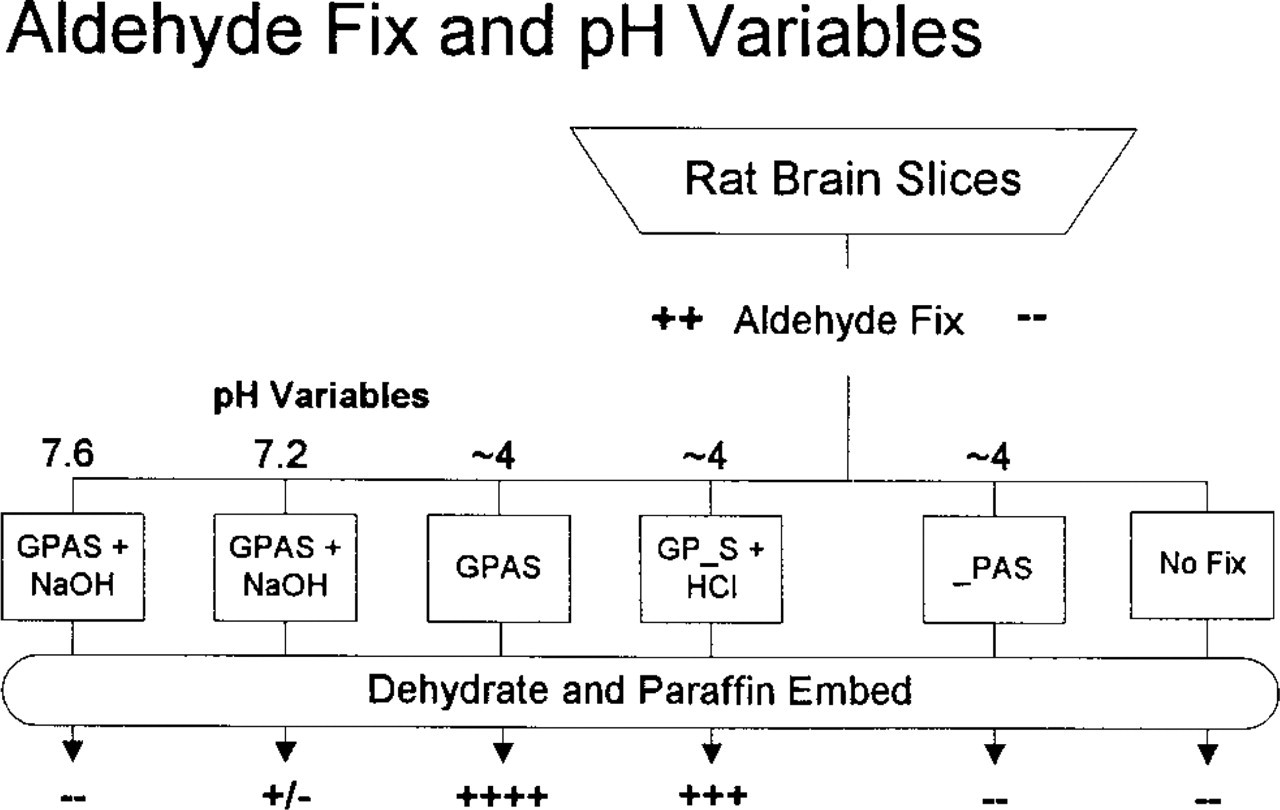
Brain slices were subjected to fixation variables for comparison with optimal LC1-ir preservation (++++) by GPAS at pH 4 (center). No LC1-ir was detected without fixation or without aldehyde (right). Raising the pH of GPAS fixative with sodium hydroxide (left) diminished (pH 7.2) or eliminated (pH 7.6) LC1-ir. However, replacement of acetic acid with hydrochloric acid (GP_S + HCl) preserved LC1-ir well.
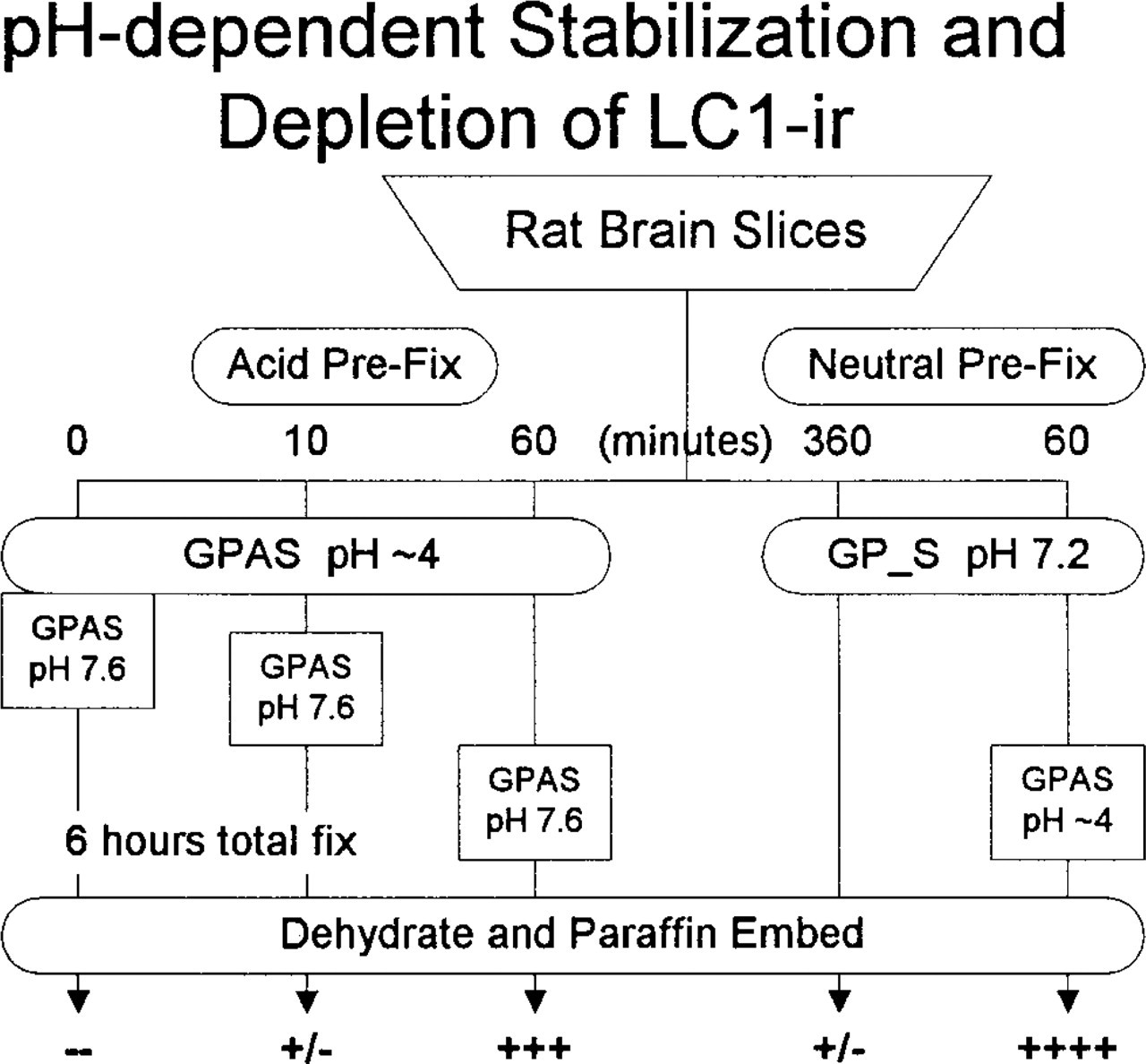
Effects of alkaline fixatives pre- and postfixation with GPAS. Alkaline GPAS was unable to preserve any LC1-ir (left). Ten min of acid GPAS pre-fixation provided minimal protection (-/+); 60 min of GPAS (center) was adequate to resist degradation of LC1-ir by alkaline fixative (+++). Neutral/alkaline destruction is not irreversible (right). After 60 min of GP_S at pH 7.2, 5-hr fixation with GPAS preserved excellent LC1-ir (++++).
Aldehydes. Because aldehyde fixation reactions reportedly are retarded under acidic conditions and accelerated under basic conditions (Berod et al. 1981), we hypothesized that the loss of LC1-ir under more alkaline conditions was due to vigorous crosslinking and that the preservation of LC1-ir by FPAS depended on suppression of fixation reactions at low pH. To test these hypotheses, we investigated the time course for loss of LC1-ir in neutral/basic fixatives (Figure 3).
Slices were pre-fixed with GPAS for 0 (control), 10, or 60 min, transferred to alkaline GPAS (raised to pH 7.6 with 1 N NaOH), and fixed for 6 hr, followed by paraffin embedding and processing for immunohistochemistry. The hypotheses predicted that LC1-ir would be absent from all three samples because, if aldehyde reactions were inhibited during the low-pH episode, then the important epitopes of the antigen would be available for destruction/blocking on transfer to pH 7.6.
The results were not consistent with the hypotheses. Fixation at pH 7.6 destroyed LC1-ir in the control (Figure 1D, top) and also in specimens that received only 10-min GPAS treatment. However, exposure to GPAS at pH 4 for 60 min preserved good LC1-ir (3+) despite subsequent treatment with pH 7.6 fixative for 5 hr (Figure 1D, bottom). Therefore, postfixation with glutaraldehyde at pH 7.6 eliminated LC1-ir from lightly fixed rat brain but did not touch it after 1 hr of fixation at pH 4. These results demonstrated that pre-fixation with aldehydes at low pH preserved LC1-ir and therefore indicated that the fixative reacted with the antigen under acidic conditions.
To test whether crosslinking at neutral/basic pH destroyed the LC1 antigenicity, slices were pre-fixed in neutral glutaraldehyde followed by GPAS at pH 4. The hypothesis predicted that the pre-fixation would seriously diminish LC1-ir. Control slices fixed in GP_S (pH 7.2) for 6 hr showed little LC1-ir (+/-) (Figure 1B, top); sister slices that spent sixty minutes in GP_S before transfer to GPAS for 5 hr displayed excellent LC1-ir (4+) (Figure 1B, bottom). Therefore, pre-fixation at neutral pH did not appear to destroy or irreversibly crosslink LC1, but acid fixation was required to preserve LC1 immunoreactivity.
If preservation of LC1-ir depended on suppression of formaldehyde reactivity at low pH, then fixation with aldehydes that are more reactive under acidic conditions should diminish LC1-ir. We tested glutaraldehyde, which reacts strongly at pH 4 (Bowes and Cates 1966), and acrolein (H2C=CH-CHO), which has been described as “a toxic aldehyde fixative that penetrates and fixes tissues rapidly” (Larsson 1988). At pH 4, essentially uniform excellent preservation of LC1-ir (4+) was obtained with 10% formalin (3.7% formaldehyde with 1.5% methanol), 2.5% glutaraldehyde (either EM grade or years-old refrigerated bulk 25% biological grade), and acrolein (2.5% EM grade from a fresh ampule). Therefore, aldehyde fixation at low pH apparently was effective.
Acid-Alcohol. Because LC1-ir preserved with formaldehyde (4%) prepared fresh from paraformaldehyde was slightly less dense (3+) than that preserved by 10% formalin (3.7% formaldehyde + ~1.5% methanol), alcohol was a variable of potential interest in fixatives. Traditional formulations of acid-alcohol (1–5% acetic acid + 70% ethanol) were negative or equivocal (+/-) for LC1-ir. Quite unexpectedly, however, lower concentrations of alcohol were effective. E20-AS (20% ethanol + 1% acetic acid + 0.9% salt) adequately preserved LC1-ir (3+) (Figure 4). Furthermore, the acetate was unnecessary; E30-HCl (30% ethanol taken to pH ~4 with HCl) also preserved LC1-ir (3+). As shown in Figure 1C, raising the acetic acid to 5% (E20-A5S = 20% ethanol + 5% HAc + salt) gave even better preservation of LC1-ir (4+).
Although acid-alcohol fixatives preserved the LC1-positive cells and LC1-ir, they were far from ideal. The ground substance of these tissues was washed out and other antigens of interest (e.g., S100-β, phosphotyrosine) were not preserved. Alcohol in the fixative did not substantially augment the immunoreactivity preserved by acidified aldehydes [adding 5–50% alcohol to F/GPAS did not change the expected maximal LC1-ir (4+)]. Furthermore, alcohol did not reverse the loss of LC1-ir with neutral/basic aldehyde fixatives.
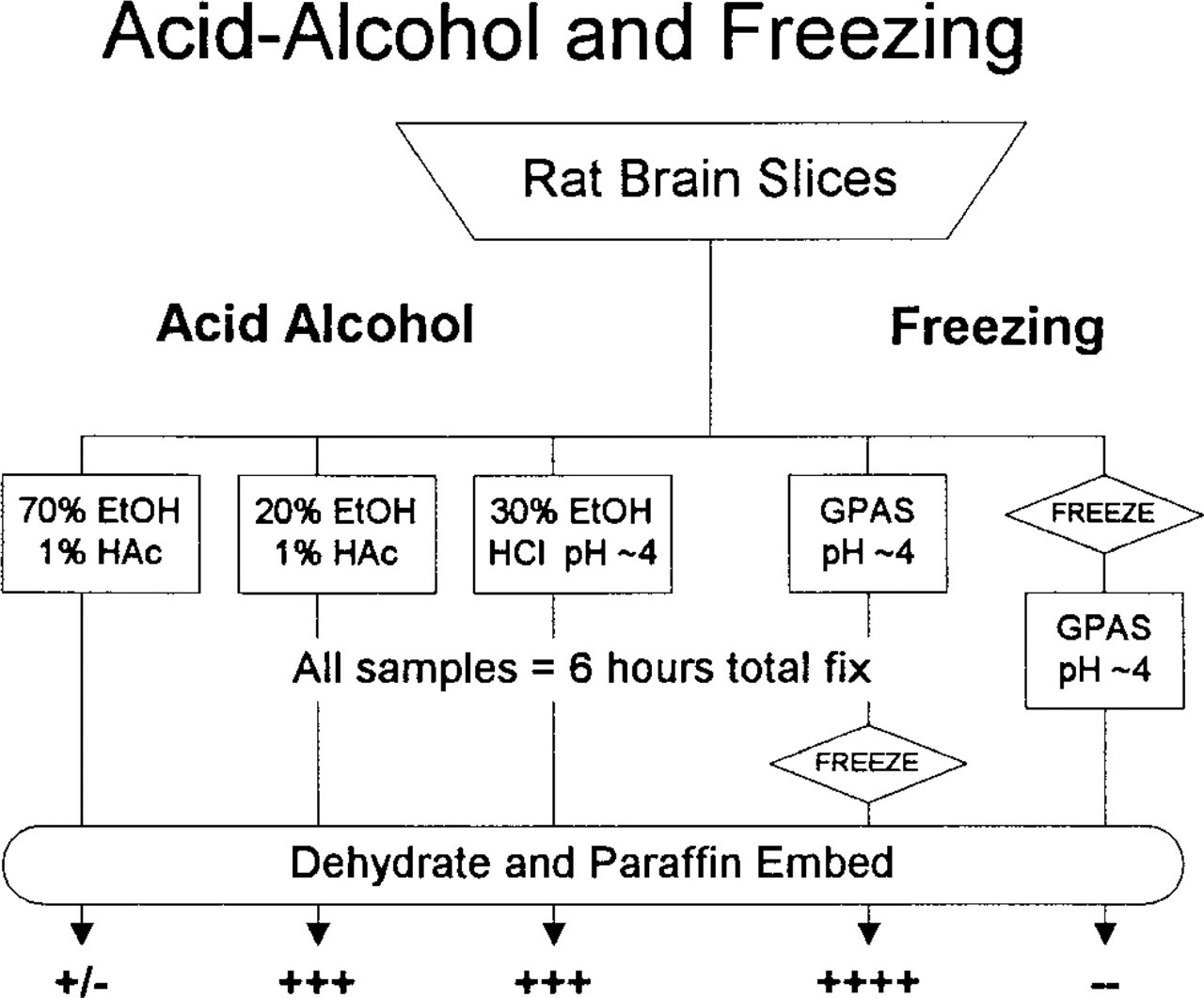
LC1-ir is preserved by primary fixation with 20–30% ethanol acidified to pH ~4 with either acetic or hydrochloric acid (center) but not by similarly acidified 70% ethanol (left). Freezing has no effect on LC1-ir after fixation with GPAS, but freezing before fixation completely eliminates LC1-ir (right).
Freezing Pre/Postfixation. Because low-affinity LC1-ir sites (demonstrable only with primary sera diluted 1:100) have been reported on neurons and astrocytes in cryosections of rat brain, we investigated the effects of freezing on LC1 immunolocalization (Figure 4). After fixation for 6 hr in GPAS or FPAS, slices were frozen, thawed, embedded in paraffin, and processed for LC1 IHC. Such pre-fixed slices displayed excellent LC1-ir (4+), demonstrating that freezing in itself does not destroy or translocate microglial LC1-ir. However, when slices were frozen before fixation (equivalent to preparing unfixed tissue for cryosectioning), LC1-ir was entirely absent (-).
To evaluate the time course of antagonism between freezing and fixation, slices were immersed in GPAS and allowed to remain at room temperature (RT) for staged periods of pre-fixation before freezing. The effects of pre-fixation on microglial staining were dose-dependent (Table 1). Somewhat surprisingly, however, staining of neurons and/or astrocytes was not detected in these experiments, even at serum dilutions of 1:100.
Cryosections
Hypothesizing that paraffin embedding might interfere with the low-affinity staining, we next examined cryosections. To confirm that LC1-ir survived the sucrose cryoprotection and freezing, residual blocks from the cryostat were thawed, embedded in paraffin, and stained. The levels of LC1-ir detected in these residual blocks (4+) were indistinguishable from control specimens that had been embedded in paraffin immediately after fixation. Furthermore, when the cryosections were stained with our standard protocol (see Materials and Methods), the serum at 1:8K dilution demonstrated strong (4+) LC1-ir (Figure 5A). Attempts to reproduce the low-affinity staining in these cryosections using primary serum at 1:100 dilution were negative; microglia were still the only cells showing DAB reaction product.
Effects of GPAS fixation before freezing: paraffin-embedded slices a
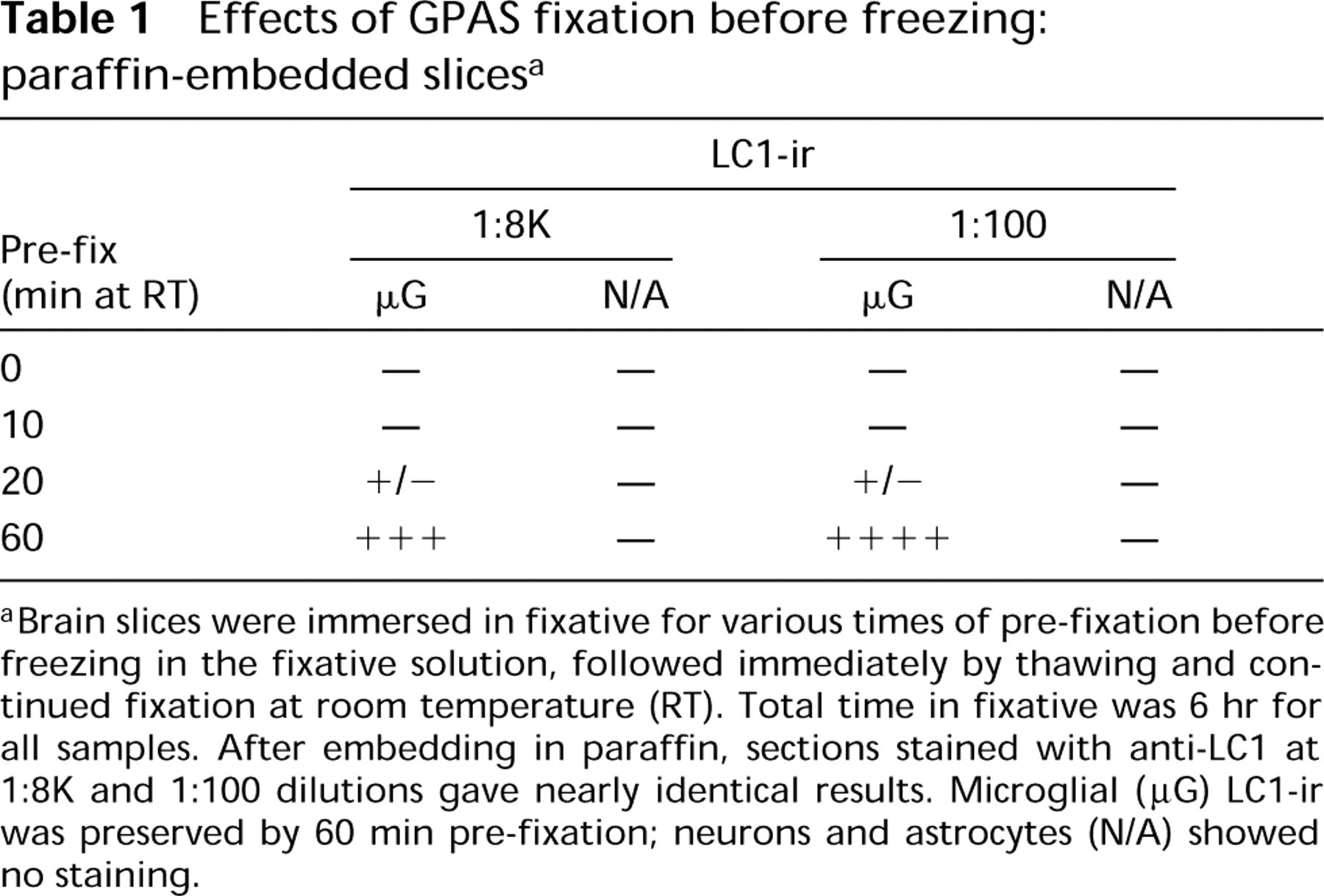
Brain slices were immersed in fixative for various times of pre-fixation before freezing in the fixative solution, followed immediately by thawing and continued fixation at room temperature (RT). Total time in fixative was 6 hr for all samples. After embedding in paraffin, sections stained with anti-LC1 at 1:8K and 1:100 dilutions gave nearly identical results. Microglial (μG) LC1-ir was preserved by 60 min pre-fixation; neurons and astrocytes (N/A) showed no staining.
The low-affinity staining had been demonstrated with fluorescence methods that did not employ the graded DeRe steps of our standard protocol. Therefore, we investigated De Re as a variable in the preservation of LC1-ir. Cryosections of FPAS-fixed tissues taken directly to primary serum at 1:8K dilution (without DeRe) were almost devoid of staining, showing only equivocal LC1-ir (+/-) in microglia (Figure 5B). Transferring the specimen directly to absolute methanol (with or without H2O2), as often done for cryosections, improved LC1-ir only slightly (+). Full development of microglial LC1-ir appeared to required DeRe through a graded series of alcohols.
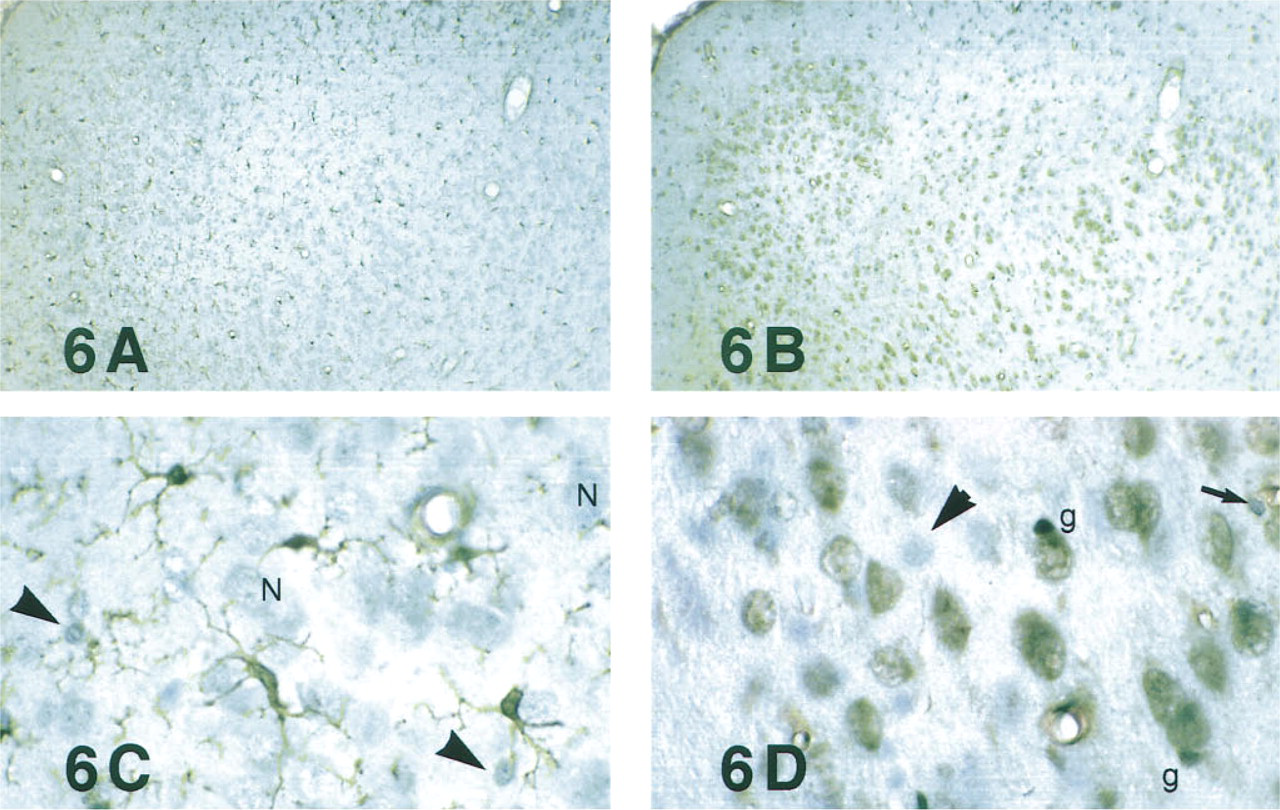
Neuron Staining. Because high-affinity staining apparently required exposure of the epitope to a hydrophobic environment, we investigated the influence of this factor on low-affinity staining. Without DeRe, cryosections displayed prominent neuron staining (3+) with primary serum at both 1:100 and 1:1K (Figures 6B and 6D). It was reduced (2+) at 1:4K (Figure 7) and absent at 1:8K (-) (Figure 5B). As shown in Figure 8, without DeRe before primary serum, neuron staining always was apparent regardless of the quality of fixation. With DeRe, neuron staining was never observed in adequately fixed brain. Cryosections of unfixed or poorly fixed brain (immersed in neutral formalin for a few hours) displayed some neuron staining (+) even after DeRe.
Microglial staining showed an inverse but less rigorous dependence on DeRe (Figure 8). Even without alcohol exposure, strongly fixed (GPAS) specimens displayed partial staining of both microglial somata and neurons (3+); the microglial ramified processes were only lightly stained (Figures 6B and 6D). Adjacent cryosections treated briefly with 30% ethanol before primary serum displayed strong microglial LC1-ir (4+) but no neuronal staining (-) (Figures 6A and 6C).
These data indicated that total dehydration was not necessary, that 30% ethanol for 15 min abolished neuronal staining, and that 10% ethanol did not. Immersion in 100% ethanol abolished neuronal staining and improved microglial staining; 100% acetone improved staining of microglial processes but did not eliminate neuronal staining. Either 30% ethanol alone or series DeRe maximized the microglia/neuron stain ratio; either was better than direct immersion in absolute ethanol.
The results with alcohols clearly showed the preponderance of microglial LC1-ir and the absence of neuronal staining in brain sections embedded in paraffin (exposed to ethanols in two steps). It was tempting to conclude that partial dehydration unmasked true LC1 epitopes in microglia and that neuron staining was unspecific. However, such conclusions required better evidence about the specificity of our immuno-reagents.
Staining Specificity
In pre-absorption controls for staining specificity, the primary antiserum is incubated with purified antigen to neutralize specific immunoglobulins before immunostaining; specific immunoreactivity is reduced or eliminated. To enhance the credibility of the results, it is important to maximize the sensitivity in these experiments. We used primary serum at 1:4K, the maximal dilution that still provided unambiguous neuron staining (2+). That is, if the purified antigen could not eliminate staining at this dilution, it never would. In addition, cryosections of GPAS-fixed brain were stained without DeRe, i.e., the protocol conducive to both microglial and neuronal staining (Figures 6B and 6D).
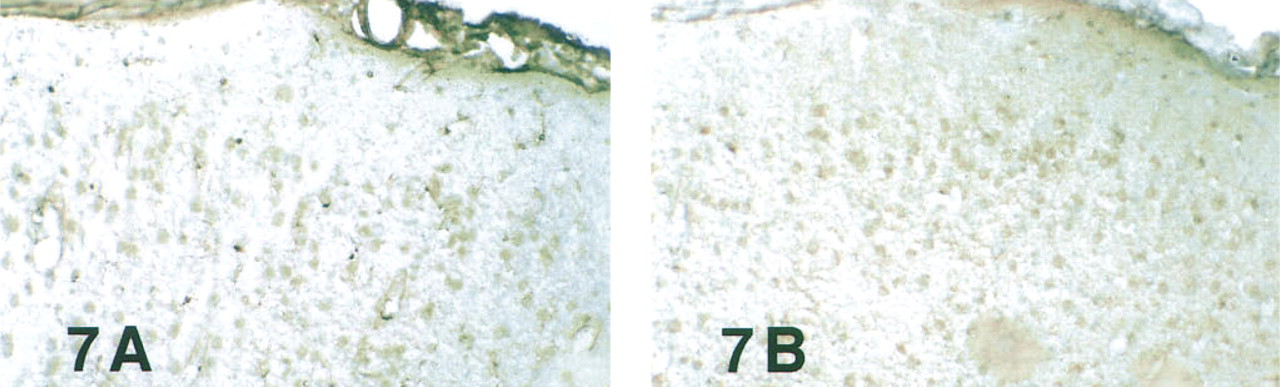
As expected, pre-absorption of anti-LC1 serum (1:4K) with 100 μg/ml bovine serum albumin did not diminish staining of neuronal nuclei or microglia (Figure 7A).
Pre-absorption with Purified Placental LC1. As shown in Table 2, pre-absorption of primary serum with placental LC1 at 1 μg/ml diminished microglial staining; 10 or 100 μg/ml totally eliminated microglial staining, but neuron staining was not reduced in any of these specimens (Figure 7B). Because the neuron staining could not be abolished by purified antigen, it should be defined as immunologically unspecific. Identical neuron-associated DAB reaction product also appeared when never-dehydrated cryosections were “stained” with normal rabbit serum.

De/rehydration, primary serum (1:8K). Cryosections (20 μm thick) cut from rat brain perfused with weaker fixative (4% paraformaldehyde + 1% glacial acetic acid) were stained as floating sections using anti-LC1 at 1:8000 dilution. (
DeRe, primary serum (1:1K). Cryosections (10 μm thick) cut from rat brain perfused with strong fixative (GPAS) were mounted on slides and stained with anti-LC1 serum at 1:1000 dilution. Section receiving DeRe treatment before primary serum (
Pre-absorption controls. Cryosections received the same treatment as in Figures 6B and D, except that primary serum diluted 1:4000 was pre-absorbed with 100 μg/ml of either bovine serum albumin (
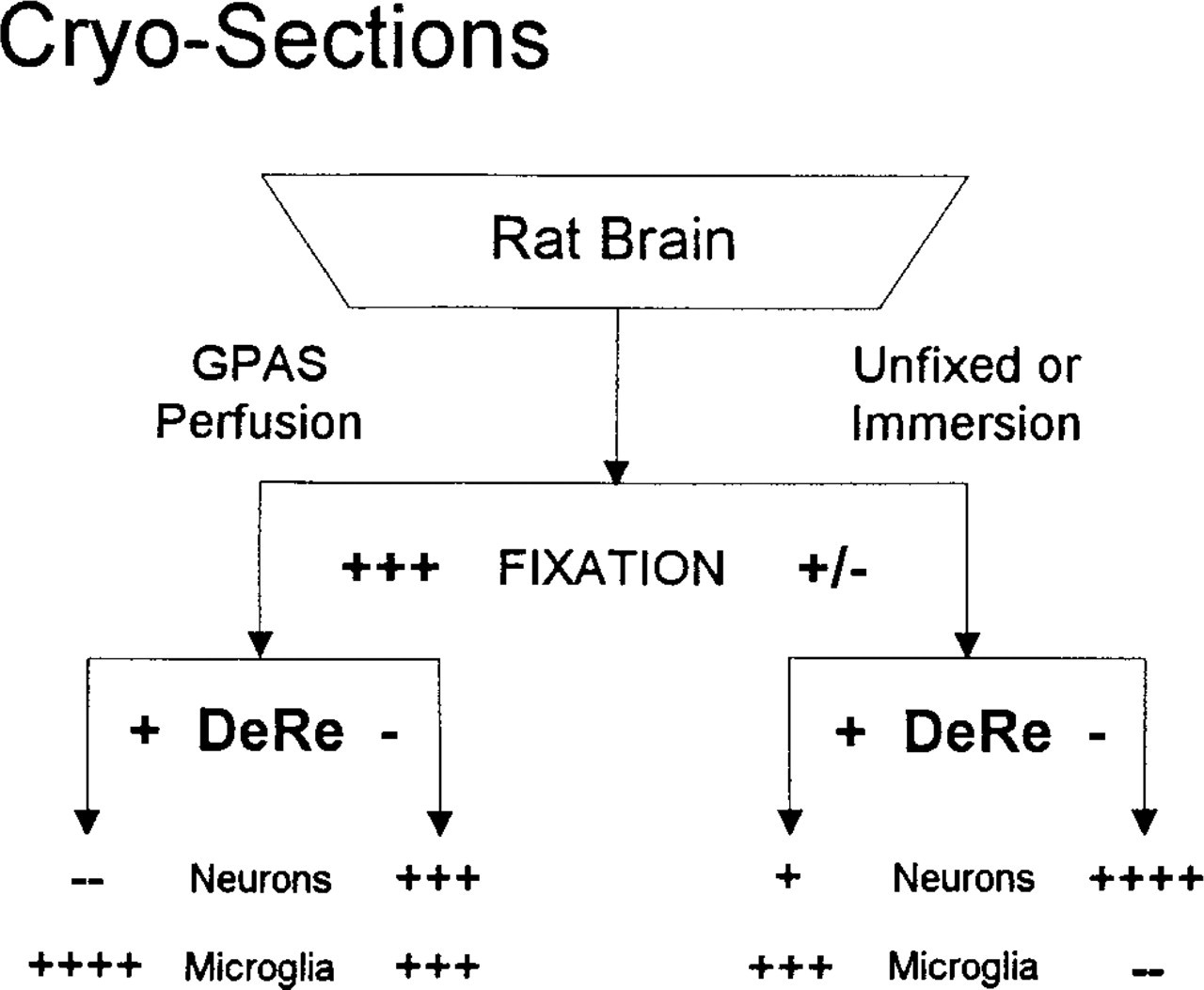
Fixation and alcohol exposure before primary antiserum (DeRe) significantly influence apparent immunolocalization of LC1 in rat CNS. In fixed brain (left), unspecific neuron staining is eliminated by DeRe; microglia stain regardless of DeRe. In unfixed or lightly fixed specimens (right), neurons stain slightly even with DeRe. Specific LC1-ir in microglia is essentially the inverse of neuronal LC1-ir, optimized by both good fixation and DeRe.
The dose-dependent reduction in microglial LC1-ir produced by pre-absorption with purified LC1 indicated specific immunoreactivity. However, these data were not conclusive. The real antigen, a purified preparation of LC1 from human placenta, contained contaminating proteins. These contaminants would raise antibodies that could recognize homologues in other mammalian specimens and result in unwanted specific staining. One approach to solving this problem was to pre-absorb with immunologically pure LC1.
Pre-absorption with Recombinant LC1. The concept of immunological purity defines a protein preparation that has very low probability of containing contaminants related to contaminants in the original antigen preparation. Specifically, experiments were designed using LC1 protein synthesized in yeast transfected with the cDNA for human LC1. Regardless of the final purity of the recombinant LC1 (rLC1) in these preparations, all contaminants would be from yeast rather than from mammalian proteins. Although not absolutely certain, it is unlikely that the yeast proteins would neutralize immunoglobulins raised against contaminating proteins from human placenta.
Pre-absorption controls a

Anti-LC1 serum was diluted 1:4K with PBS containing 10% goat serum, incubated overnight with different concentrations of purified placental LC1, and used to stain cryosections from well-fixed brain (perfused with GPAS).
For further assurance, we tested rLC1 from two quite different strains of transfected yeast, Saccharomyces diastaticus (Nam et al. 1994) and Schizosaccharomyces pombe (Tohda et al. 1994). With both strains of yeast, the results were identical. Pre-absorption with the Ca++-sensitive protein fraction containing rLC1 reduced and eliminated LC1-ir from microglia in a dose-dependent fashion. Control protein preparations from parent strains (either wild-type or transfected with human non-annexin proteins) had no effect on LC1-ir in microglia. Neuron staining in cryosections was not reduced by pre-incubation with rLC1.
Solid-phase Pre-absorption The pre-absorption controls using yeast protein fractions enriched for rLC1 provided strong evidence that neuronal staining was unspecific. However, we were concerned that similar results regarding elimination of microglial staining would be observed if the rLC1 fraction simply destroyed the anti-LC1 immunoglobulins. Perhaps protease activity or other destructive/masking factors from yeast contaminated the rLC1 fraction.
To test this hypothesis, yeasts (fixed and permeabilized) were used as solid-phase substrates for immobilized antigen in a combined pre-absorption control and affinity-purification protocol (see Materials and Methods). Aliquots of anti-LC1 serum at 4 × working dilution (1:2K) were mixed with equal volumes of control and LC1 yeast. After 60 min for binding equilibration, the tubes were centrifuged and the supernatant used for immunostaining at a final dilution of 1:8K. Sera pre-absorbed with control (wild-type or parental) strains produced full undiminished LC1-ir (4+), whereas those pre-absorbed with recombinant yeast were weak to nil (+/-). Therefore, the pre-absorption results with intact yeast were identical to purified rLC1, but these data did not eliminate the possibility that the anti-LC1 was removed by lytic rather than immunospecific factors.
Affinity-purified Anti-LC1. If factors in the yeast had destroyed anti-LC1 immunoglobulins, then the recovery of immunoreactive IgGs from the yeast would be impossible. On the other hand, if the IgGs bound specifically to LC1 in the yeast, they would remain bound through repeated washing and could then be eluted and used to stain microglia. After repeated washing of the yeast, a single acid rinse released bound antibodies from the immobilized antigen. The acid rinse was immediately neutralized and used for immunohistochemistry. The solutions recovered from washed control yeast showed no LC1 immunoreactivity (-); those from rLC1 yeast stained microglia very well (3+). These results provided strong confirmation that the IgGs staining microglia recognize human LC1.
Discussion
Immunochemistry of Lipocortin
As reviewed by Larsson (1988), certain criteria are generally recognized as hallmarks of immunohistochemical specificity. These include the use of high-titer antisera at great dilution, total loss of staining when antisera are pre-incubated with purified antigen, corresponding immunolocalization and immunoblot data on tissue distributions, and consistent levels of immunoreactivity under varied conditions of fixation, sectioning, and staining.
Our studies of variables that influence detection of LC1-ir have largely satisfied the criteria above. With appropriate tissue fixation, LC1-ir is stained with antisera raised against LC1 purified from different species, including hog and human. These polyclonal antisera are effective at 1:8000 dilution, they identify LC1-ir in identical cell types in both frozen and paraffin sections, and the staining is completely abolished by pre-incubation with purified LC1. Studies in kidney have shown that pathophysiological changes in levels of LC1-ir quantified in tissue sections corresponded precisely with levels measured from immunoblot (Western) analysis (McKanna et al. 1992).
The lipocortin antigen has some unusual characteristics. For example, freezing tissues before fixation totally eliminates LC1-ir and fixation of LC1-ir requires acidic conditions. Dilute acid-alcohol preserves LC1-ir but it is diminished by more concentrated alcohols. Finally, partial dehydration is required for immunodetection. Therefore, preservation and detection of LC1-ir appear to be sensitive to pH and hydrophobicity.
Studies in other disciplines have reported physiological and physical chemical properties of LC1 that appear relevant to its unusual immunohistochemical characteristics. Although it is traditionally described as a soluble cytoplasmic protein, LC1 has unusual sedimentation and partitioning properties. Early studies showed that LC1 binds to and sediments with membranes and cytoskeleton in the presence of Ca++ and that it is released into the supernatant when cell homogenates are treated with Ca++ chelating agents (Ahn et al. 1988; Fava and Cohen 1984). Subsequently, however, a Ca++-independent fraction of LC1 was discovered. Approximately 50% of the LC1 in human placenta becomes soluble when Ca++ is chelated in homogenates, but the other half cannot be solubilized without detergents and presumably is complexed with membranes (Myatt et al. 1992). Furthermore, annexins reportedly behave as partially lipophilic proteins, partitioning to acidic chloroform-methanol but showing little affinity for the organic phase at neutral pH (Genge et al. 1991).
We postulated that the influence of pH and alcohol on preservation and detection of LC1-ir was related to some or all of these physicochemical properties. However, preliminary attempts to test these properties directly yielded negative results. Extraction of brain slices with acidic chloroform-methanol for 30 min before GPAS fixation destroyed tissue integrity but only partially reduced LC1-ir (2+). Incubation of brain slices in PBS + EDTA (5 mM) for 2 hr before fixation caused no detectable diminution or translocation of LC1-ir (4+).
Lipocortin in the Central Nervous System
There is little question regarding the presence of LC1 in rat central nervous system (CNS). It was detected by both immunoblotting and PLA2 inhibitory activity (Rothwell and Relton 1993; Regnouf et al. 1991). In adult rat brain, LC1 levels reportedly are stable under control conditions and are elevated after disease or experimental trauma (Eberhard et al. 1994; Williams et al. 1994; McKanna 1993a; Elderfield et al. 1992).
Several questions regarding LC1-ir in the CNS cannot be resolved at present. Both species and age differences are apparent. In some cases the immunoreactivity is robust. With current antisera, an LC1-positive midline raphe is detected in the floor plate of vertebrates ranging from primates to zebrafish (McKanna 1993b; and unpublished observations). However, LC1-ir in the floor plate raphe is preserved by almost any acidic fixative, e.g., Carnoy's, Bouin's, or 70% ethanol + 30% acetic acid (Hamre et al. 1995; McKanna and Cohen 1989).
In other cases LC1-ir appears elusive. Strong LC1-ir is detected in mouse microglia in culture (Fedoroff and McKanna 1994) but, regardless of fixation variables, LC1-ir is never observed in resting ramified microglia of adult mice. Further study may indicate whether the various sites and species have different isoforms of LC1 or whether the different staining patterns are due to downregulation of LC1.
In contrast, an overabundance of LC1-positive cell types has been reported in rat CNS. LC1-ir was localized to GFAP-positive reactive astrocytes in lesioned rat cerebellum cryosectioned without fixation or cryo-protection (Mullens et al. 1994). We have been unable to demonstrate LC1-ir in fresh frozen rat brain under any circumstances. We also have shown that the high levels of LC1-ir in salivary gland ducts are not detected in tissues frozen before adequate fixation, even when the sections are immediately postfixed in GPAS (McKanna, unpublished observations). Further investigation is needed to determine which factors modulate the detection of LC1-ir in fresh frozen tissues.
In other cases, several minor problems appeared to multiply. Neutral formalin preserves LC1-ir in surface cells exposed to high levels of fixative, leading to staining of ependymal cells (Strijbos et al. 1991), but neutral formaldehyde does not preserve LC1 in the delicate ramifications of stellate glia, making cytological identification difficult. In normal rat brain, microglia can be identified by Griffonia simplicifolia isolectin B4 and by immune system markers such as Mac-1 and F4/80 (reviewed by Streit and Kincaid-Colton 1995). Our studies have shown that the LC1-positive cells comprise the populations identified by other microglial markers (McKanna 1993b) and that, in response to several different types of CNS lesions, these LC1-positive microglia proliferate and become phagocytic. These same studies demonstrate that the population of LC1-positive cells is quite distinct from astrocytes identified in our specimens by S100-β immunoreactivity. The astrocytes participate little in reactive gliosis (McKanna 1993a). Furthermore, we have demonstrated in culture that microglial progenitors express LC1 and are distinct from astrocyte progenitors from the earliest stages of morphogenesis (McKanna and Fedoroff 1996; Fedoroff and McKanna 1994). Therefore, LC1 appears to be a comprehensive and reliable marker for microglia.
pH and Fixation
Evidence that acidic fixation conditions favor preservation of LC1 and several other small, soluble protein antigens leaves two histological points open to reconsideration. First, the tradition of fixing tissue near physiological pH may be irrelevant, since the goal is to fix/immobilize/denature the biological molecules. It may be helpful to use conditions that change the charges and conformations of these molecules and inhibit their activities. Second, although the reactions of formaldehyde with collagen are accelerated at pH 7<9<11 (Berod et al. 1981), we are unable to distinguish structurally between tissues fixed at pH 4 and those fixed at neutrality. This is even more true in the case of glutaraldehyde, for which the speed of fixation appears to be approximately half-maximal at pH 4 (Bowes and Cates 1966).
Because both FPAS and GPAS provide excellent preservation, both have been useful in investigations of true LC1-ir localization. It is worth mention, however, that the preservation achieved with GPAS or FPAS is not equivalent for all antigens. GPAS significantly enhances preservation of phosphotyrosine (PY-ir) and cyclo-oxygenase-2 (COX-2) (Harris et al. 1994; McKanna and Casagrande 1994); FPAS is preferred for heparin-binding EGF (Sakai et al. 1995).
In summary, the requirements for fixation and immunolocalization of LC1 are specific and unusual. A variety of aldehydes are acceptable for preserving LC1-ir, hydrophobic and acid environments expose critical epitopes, and LC1-ir is very sensitive to freezing. Attention to these details may help to resolve conflicting data regarding the tissue and cellular distribution of LC1 and may contribute to future investigation of its functions.
Footnotes
Acknowledgements
Supported by NIH grants NS32660, HD15052, and DK39261, and by ONR FEL Center N14-94-1-1023.
We thank Natalie Ahn and Stanley Cohen for antisera, Bong Hyun Chung and Yuko Giga-Hama for recombinant yeast, Michael Bienkowski for unpublished data on LC1 purification, and David Lovinger and Elizabeth Tyler for brain slices.