
Abstract
Biotin was recently applied to detect cellular DNA or RNA. In combination with avidin, streptavidin or antibody, it can be conjugated with fluorescent dye, enzyme, ferritin, or gold. However, emphasis has recently been placed on the false-positive results that are obtained when this probe is used, because endogenous biotin may sometimes interfere with specific signals. Digoxigenin appears to be an interesting alternative because it is present exclusively in Digitalis plants as a secondary metabolite. We discuss in this review the efficiency and the respective advantages and disavantages of these two probes for in situ hybridization, mainly at the electron microscopic level.
Keywords
The introduction in the late 1960s of in situ hybridization (ISH) techniques (Buongiorno–Nardelli and Amaldi 1970; Gall and Pardue 1969; John et al. 1969) opened a new era in histology and cell biology. Whereas immunocytochemical methods can demonstrate only the presence of synthesized protein molecules, irrespective of any routing in the tissue, the recognition in a tissue and in a cell of specific DNA or RNA sequences defines the precise location of a potential or an effective synthesis of a given molecule. ISH comes from the techniques of molecular hybridization of nucleic acids that are isolated from a particular cell population or tissue and bound to solid supports. Whereas hybridization of such averaged membrane bound nucleic acids can identify different classes of DNA (Southern blot) and RNA (Northern blot), ISH fills the gap between the detection of a specific sequence and its precise location within the tissue or the cell. A major advantage of ISH is that it allows literally hundreds of different hybridizations because it is often performed on thin or ultrathin sections of a piece of tissue (e.g., a single surgical biopsy) which, at times, is not sufficient to allow Northern or Southern blot analysis. It is also possible to make libraries of paraffin- or resin-embedded or frozen tissues. No significant loss of the hybridization signal was found in frozen sections kept at − 70C with dessicant for more than 6 years (Wilcox 1993).
The sensitivity and efficiency of ISH depend on several variables, for which optimal conditions must be determined: (a) the probe construction and hybridization conditions; (b) the type and efficiency of probe labeling; (c) the tissue preparation (fixation, embedding) which must allow the retention of the target of hybridation and/or the hybridized products; and (d) the method used for signal detection.
We discuss in this review the efficiency and the respective advantages and disavantages of biotin and digoxigenin as labels for in situ hybridization probes, mainly when used at the electron microscopic level.
The Emerging Interest in Non-radioactive Probes Labeled with Biotin or Digoxigenin for In Situ Hybridization
Radioactively labeled DNA or RNA probes, as originally used in 1969 by Gall and Pardue and John et al. (1969), are still widely applied for ISH because of their high sensitivity and the amplificatory effect of autoradiography. Signal detection can be achieved with autoradiography employing liquid emulsion, the exposure time depending on the radioisotope used for labeling [3H-labeled probes require usually a rather long exposure (weeks) whereas 35S-labeled probes can yield autoradiographs within days], specific activity of the probe, copy number of target DNA or RNA, efficiency of hybridization, and sensitivity of the detection system (liquid emulsion). Safety problems, reduced stability of radioactively labeled probes, and speed of visualization as well as the extensive development of immunogold cytochemistry, which allows a much more precise location of antigen sites in a tissue section compared to the autoradiographic technique, have stimulated interest in the development of nonradioactive probes (Van der Ploeg et al. 1986; Baumann 1985; Forster et al. 1985).
Direct immunofluorescence microscopic hybridocytochemistry, applying fluorochrome-labeled DNA or RNA (Baumann 1985), is not widely used because of its relatively low sensitivity. Other developments based on immunohistochemical detection of chemically modified nucleic acids, such as acetylaminofluorene (Landegent et al. 1984; Tchen et al. 1984), dinitrophenyl (Dnp) groups as a hapten (Shroyer and Nakane 1983), and mercurated probes and sulfhydrilhapten ligands (Hopman et al. 1986), are promising. The immunohistochemical localization of DNA-RNA or RNA-RNA hybrids by specific antibodies directed to these (nonphysiological) constructs was described by several groups (Raap et al. 1984; Van Prooijen-Knegt et al. 1982; Rudkin and Stollar 1977).
The development in 1974 (Heitzmann and Richards 1974) of the biotin-avidin system to detect antigens at the electron microscope level, was a significant improvement over the immunocytochemical methods. Biotin, a small vitamin molecule (Mr 244), binds with high affinity (kD 10-15 M-1) (Green 1975) to avidin, a protein largely distributed in egg whites (Mr 70,000), which can be conjugated to different markers such as fluorescent dyes, peroxidase, ferritin, and colloidal gold. With the synthesis of biotin-labeled (d)UTP, the construction of biotinylated nucleic acids became possible (Brigati et al. 1983; Hutchison et al. 1982; Singer and Ward 1982; Langer et al. 1981) and opened important new prospects in non-radioactive ISH at both the light (LM-ISH) and the electron microscopic (EM-ISH) levels. However, biotin is an endogenous molecule of living cells associated with carboxylases and plays a key role in many reactions, mainly in the liver and the kidney (Varma et al. 1994; Kirkeby et al. 1993). In some cases, this endogenous biotin can lead to false-positive results as we will discuss below. A few years ago, digoxigenin, a steroid isolated from digitalis plants (Digitalis purpurea or Digitalis lanata), was proposed as an alternative to biotin for labeling of hybridizing probes. Because the blossoms and leaves of these plants are the only natural sources of digoxigenin, no binding of the anti-digoxigenin antibody occurs in other biological material.
Pros and Cons of Biotin and Digoxigenin in Probe Construction and Hybridization
Two parameters must be kept in mind in constructing hybridizing probes. The detectable molecule introduced chemically or enzymatically (the reporter molecule) should not interfere with the hybridization reaction or the stability of the resulting hybrid. It should also remain accessible to the detection system used later on.
As shown in Figure 1, biotin or digoxigenin is linked to uridine nucleotides at the number 5 (or 3) position of the pyrimidine ring via a spacer arm whose length can vary from 7 to 20 C/N atoms. This spacer avoids steric hindrance and allows good matching of bases during the hybridization. By extending the biotin/digoxigenin moiety further from the nucleotide on the linker arm, antibody binding is optimized. In fact, because the binding site of avidin (and presumably of streptavidin) resides within a deep (1 nm) depression (Green 1975), the spacer between biotin and the nucleotide should be at least 1 nm long. In an experiment of binding of [125I]-streptavidin to biotinylated toad erythrocytes, Bonnard et al. (1984) demonstated that a 7-atom spacer was long enough to allow a significant increase in the rate of binding, and additional elongation to 16 atoms did not further increase the rate of reaction. The length of the biotin-11 atom linker (one of the most frequently used) is ~2.1–2.2 nm (Hiriyanna et al. 1988). The variety of biotinylated nucleotides has been enlarged with the synthesis of biotinylated adenosine and cytosine triphosphate (Gebeyehu et al. 1987).
The choice of probe (cDNA, RNA, oligonucleotides) depends on the final target of hybridization. In all cases, non-radioactive ISH requires 10- to 50-fold higher concentration of probes (personal observation; and Bloch 1993) than radioactive ISH, and the overall efficiency of the probes can be classified as follows: 35S > 32P > 3H ≥ biotin/digoxigenin (Wilcox 1993).
Synthetic oligonucleotides are usually enzymatically labeled by tailing of the 3′-end with terminal deoxynucleotidyl transferase (Normand and Bloch 1991; Guitteny et al. 1988). Riboprobes are generated by in vitro transcription using a linearized template and a promoter for RNA polymerase (Ozden et al. 1990; Forster et al. 1985). These single-stranded anti-sense RNAs are the most sensitive probes for target RNA (Cox et al. 1984) and the yield of preparation was high, with 10 μg of non-radioactively labeled RNA probe obtained in the standard assay when about 1 μg of input DNA was used. However, these probes are labile. Nick-translation (Langer et al. 1981; Rigby et al. 1977) and random priming (Feinberg and Vogelstein 1984) methods are commonly used to prepare biotin or digoxigenin DNA probes. Both methods allow great flexibility with regard to length of the labeled fragments. The efficiency of labeling does not depend on the reporter molecule (biotin or digoxigenin), as long as the spacer arm is sufficient to avoid steric hindrance. Random primed labeled probes are often preferred for blot and ISH applications because of the high incorporation rate of nucleotides and the high yield of labeled probe obtained: 350–600 ng of probe (100–1000 BP) is obtained by using 10 ng to 3 μg of input DNA in the random-primed labeling reaction compared to 200 ng of probe (200–500 BP) obtained by using 100 ng to 3 μg of input DNA in nick-translation. This is illustrated in Figure 2, in which we compared the efficiency of incorporation into a cDNA probe of biotin, using nick-translation, and digoxigenin, using random priming. Because of the variability of reporter incorporation, it might be useful, before performing ISH, to evaluate the incorporation of biotin or digoxigenin into the probe by a simple dot-blot analysis, similar to those shown in Figure 2, with reference to dots of standard labeled DNA.
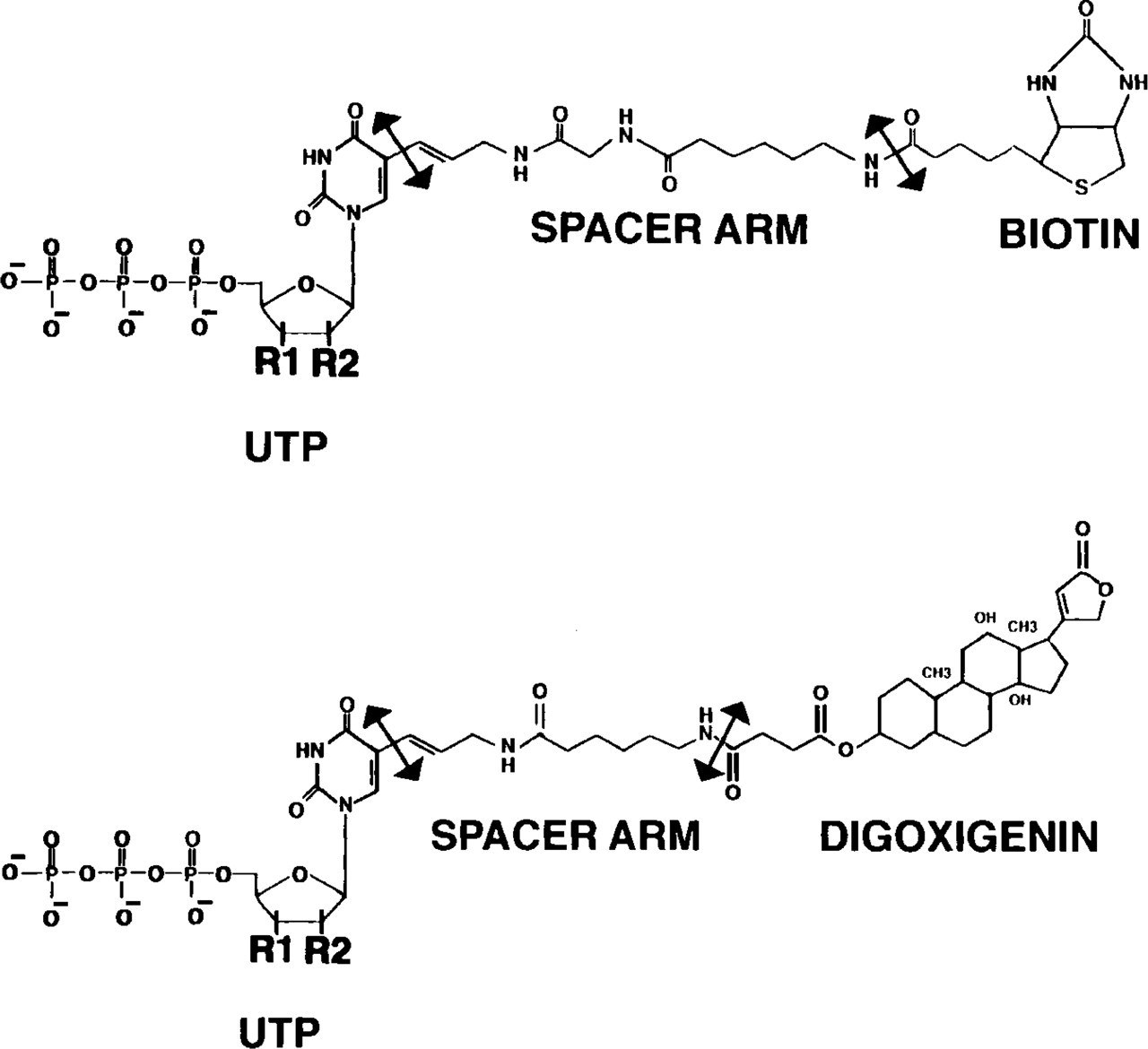
Biotin-14- and digoxigenin-11-uridine triphosphate. Biotin or digoxigenin are linked through a spacer arm whose length can vary from 7 to 20 C/N atoms, to uridine (R1 = OH; R2 = OH), deoxyuridine (R1 = OH; R2 = H) or dedeoxyuridine (R1 = H; R2 = H).
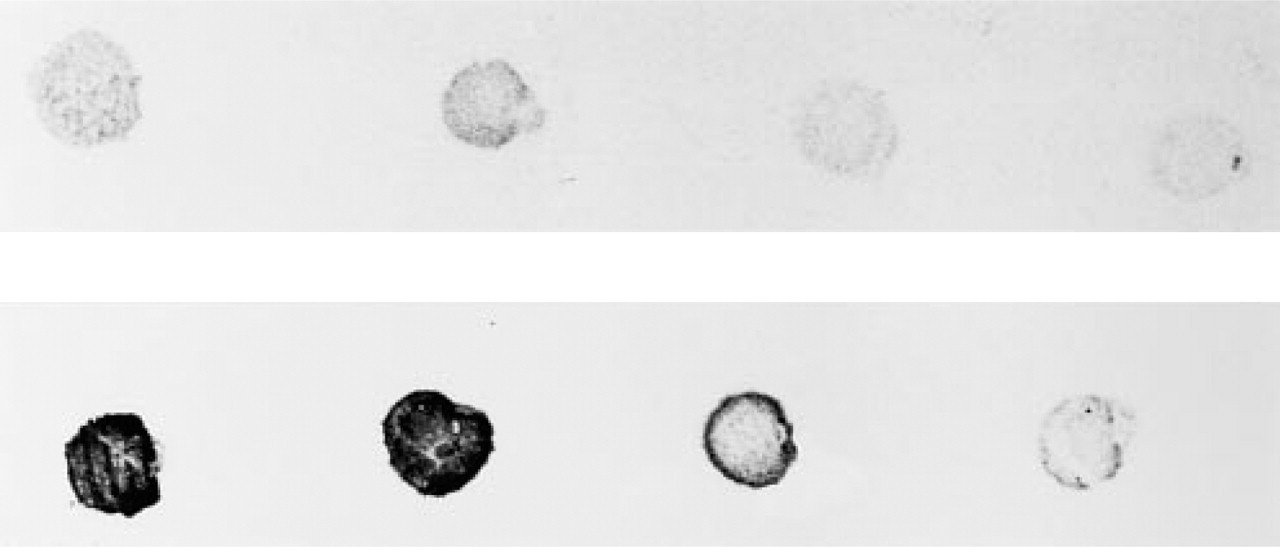
Dot-blot comparison of incorporation into cDNA probe of biotin, using nick-translation (upper lane), and digoxigenin, using random priming (lower lane). Labeled DNA dilutions were 1, 1:2, 1:4, and 1:8 (left to right). The biotin-labeled probe required more DNA (300 ng DNA) than the digoxigeninated probe (200 ng) for a lower incorporation (lighter dots).
Non-radioactive ISH is used to detect a variety of nucleic acid sequences in mature, developing, and pathologically altered tissues. Unless isolated cells are concerned, most of the time ISH must be performed on tissue sections to gain access to the deep center of the sample. The level of resolution depends on the technique used to reveal the reporter molecule and on the level of examination, light or electron microscopy.
LM-ISH is sometimes performed on sections of frozen, unfixed tissue. In this case, nucleic acid sequences are well maintained if the sample is rapidly frozen after excision of the sample; labeled probes as well as the detection systems used in the following steps easily enter the tissue. In that respect, no difference was seen between biotinylated or digoxigeninated probes, the size and the charge of the probes being roughly similar (Figure 1). However, it is clear that radioactive probes might enter the section more easily, because they do not have a spacer arm and a reporter molecule.
Frozen sections, unfortunately, retain a relatively poor structural preservation of the tissue and LM-ISH is usually developed on sections of paraffin embedded tissue, sections which are dewaxed, rehydrated and permeabilized before use (Scherthen and Cremer 1994; Han et al. 1992; Clavel et al. 1991; Brigati et al. 1983). For EM-ISH, hybridization can be done on ultrathin sections of frozen tissue, albeit only a small amount of work has depicted this approach (Le Guellec et al. 1992; Morel et al 1989a,b; Morel and Doucet 1986). Some authors have performed EM-ISH on vibratome sections (Le Guellec et al. 1992; Yun and Sherwood 1992; Manuelidis and Borden 1988) or cell suspensions (Wolber et al. 1988,1989) subsequently embedded in epoxy resin. All of these techniques allow a relative permeability to the hybridizing probe and its detection system.
LM-ISH, and especially EM-ISH, is also performed on sections of tissue embedded in epoxy or hydrophilic resins. The latter affords better preservation of DNA/RNAs than epoxy media do, because tissue preparation and embedding are performed at low temperature. Despite the fact that postembedding LM- or EM-ISH could reach all cellular substructures at the surface of the section, labeled probes can hybridize with only a few copies of the target sequences of nucleotides located at the very surface of the section. In addition, only those molecules with their longitudinal axes parallel to the surface can be hybridized and labeled (Yi et al. 1995) (Figure 3). Compared to globular antigen proteins which have several epitopes exposed at the surface of the sections of embedded tissues and thus accessible to antibodies, hybridization would not be efficient if only short ends of mRNA molecules outcrop (Figure 3). All nucleotide sequences inside the section cannot be hybridized because the probe, as well as the detection system, does not enter the section. Bendayan et al. (1984) have demonstrated elegantly that immunogold conjugates never enter the depth of a section but rather remain stuck at its surface, even when hydrophilic resins are used. Pretreatment of sections with etching reagents and digesting enzymes have been reported to improve hybridization efficiency in postembedding ISH (Morey 1995; Lin et al. 1993; Mandry et al. 1993; Puvion–Dutilleul and Pichard 1992; Puvion–Dutilleul and Puvion 1991; Puvion-Dutilleul et al. 1991; Wolber et al. 1988). However, cell structures were damaged and nonspecific binding of gold particles increased, probably owing to surface roughness of sections engendered by etching (Yi et al. 1995). The efficiency of pretreatment steps used in viral genome detection might be interpreted as the strong alkaline or acid conditions denaturing the viral DNA double helix, thereby facilitating hybridization, and structural damage is less of a concern in these settings because extranuclear morphology is irrelevant.
In conclusion, the efficiency of hybridization strongly depends on the type of probe and its construction and the type of tissue preparation, which can impede access to target DNA or RNA sequences. Biotinylated and digoxigeninated nucleotides appear to be used indiscriminantly to construct probes because they have roughly similar size and charge. However, as discussed below, it appears that the efficiency of detection of labeled hybrids might be better when digoxigeninated instead of biotinylated nucleotides are used.
Pros and Cons of Biotin and Digoxigenin in the Detection System
Because avidin is easily conjugated to ferritin and other electron dense marker or fluorescent dyes (Morris et al. 1992; Hsu et al. 1981; Guesdon et al. 1979; Heitzmann and Richards 1974), it has been extensively used in immunocytochemistry to detect biotinylated molecules. However, problems of nonspecific binding have been reported, which were attributed to the stickiness of the protein caused by its carbohydrate moieties (Hofmann et al. 1980). In addition, the pI of avidin is 10, which may favor ionic interactions with anionic cell surfaces and, hence, partially explain nonspecific binding of avidin conjugates (Wooley and Longsworth 1942). Streptavidin, released from cultures of Streptomyces avidini, has properties similar to those of avidin, i.e., both proteins are tetramers with subunit molecular weights of about 15,000, are rich in tryptophan, bind biotin with extremely high affinity, and are stable to treatment with urea, guanidine-HCl, and heat. Streptavidin is non-glycosylated and has a neutral pH (Chaiet and Wolf 1964). The latter properties make streptavidin a superior reagent for detection of biotinylated ligands.
To enhance ISH detection sensitivity of biotinylated hybridized probes, avidin or streptavidin is often used in a cytochemical network of amplifying layers such as conjugated avidin–biotinylated anti-avidin antibody–conjugated avidin (Pinkel et al. 1986) (Figure 3c). When ISH is performed on semithin or ultrathin sections, only a small part of the biotinylated probe interacts with the nucleotide sequence of the target DNA or RNA emerging at the surface of the section. The introduction of conjugated avidin or streptavidin could lead to a firmly closed network, masking overall biotin residues, which then could not be detected (Figure 3d). In such a case, the use of anti-biotin antibodies appears to be a better alternative (Figure 3e). We tested several immunogold systems for the detection of probes hybridizing to the corresponding mRNA in postembedding EM-ISH (Yi et al. 1995). The two-step approach (anti-biotin rabbit antibody/gold-conjugated anti-rabbit goat antibody) was clearly more sensitive than the one-step approach (gold-conjugated anti-biotin antibody), because no reproducible labeling was obtained with the latter. This indicates that in EM-ISH, an amplification of the signal is crucial to overcome the low number of recognizable hybridization sites.
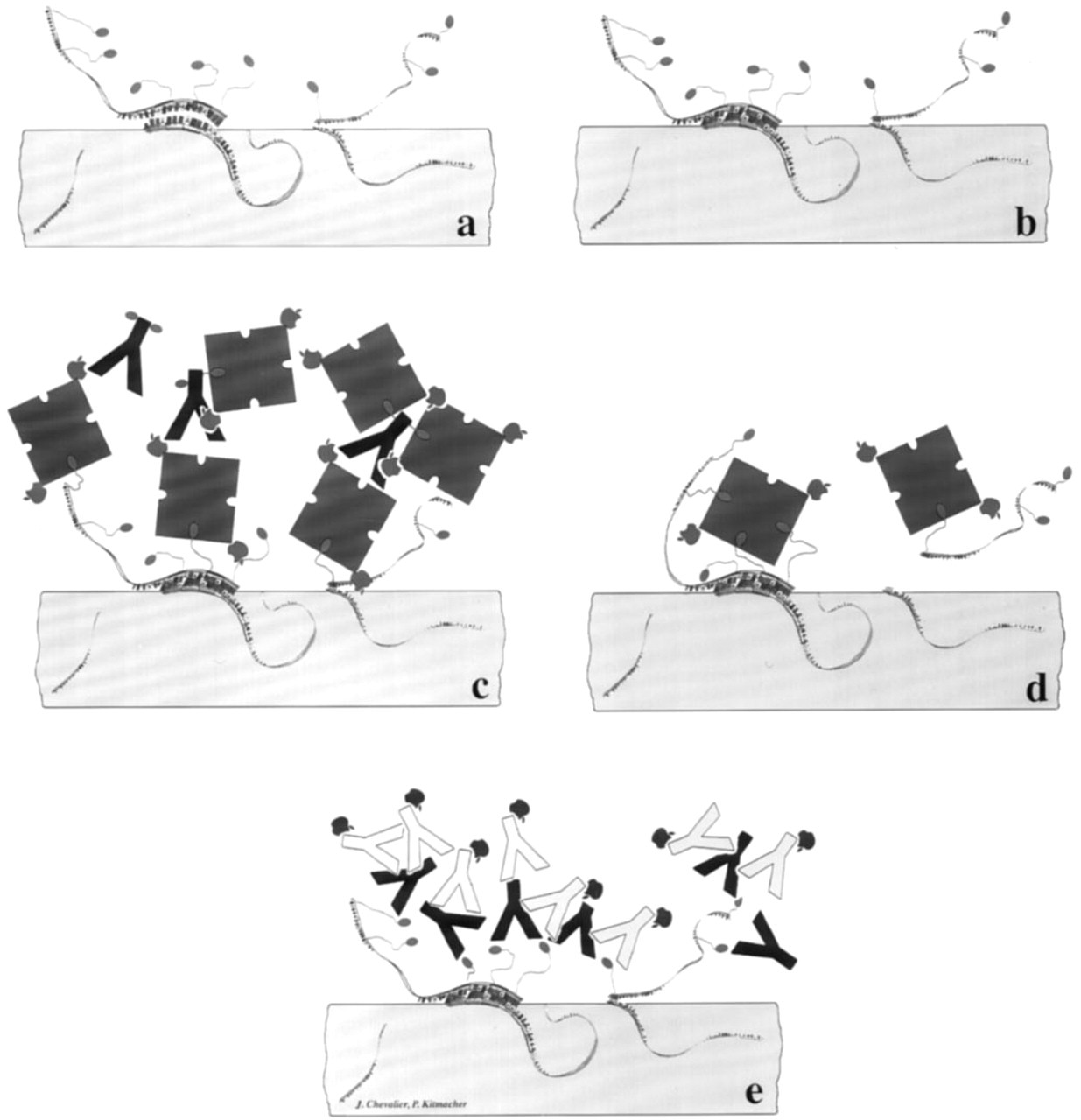
Schematic representation of hybridization of labeled probes to target RNA or DNA and recognition of the reporter molecule in postembedding ISH. Labeled probes can hybridize only with target sequences emerging at the surface of the section, but do not enter the section (
In detecting Type I collagen mRNA in rat kidneys using postembedding EM-ISH and biotinylated cDNA probes (Yi et al. 1995), we observed large amounts of gold particles on mitochondria of interstitial fibroblasts and tubule cells, in an area at which no target mRNA should be found (Figure 4). We then checked for endogenous biotin, performing a histochemical test on intact frozen tissue identical to that used for EM-ISH. The strong positive staining obtained on kidney tubule cells confirmed that endogenous biotin must be considered when biotinylated hybridized probes have to be detected. Biotin is present in the cytosol of every cell type (Varma et al. 1994; Kirkeby et al. 1993; Bianchi et al. 1990; Kuhn 1988). In certain tumor cell lines, the levels of endogenous biotin-containing enzymes have been shown to vary depending on their degree of tumoroginicity (Bramwell and Humm 1992). Interference with true hybridized signals would be hard to circumvent even with an efficient blocking reagent (Varma et al. 1994). Results obtained with biotin-based systems should therefore be interpreted with care. Although the biotin system has been used extensively (Bloch 1993; Mitchellet al. 1993; Le Guellec et al. 1991; Guitteny et al. 1989), few authors have discussed interference by endogenous biotin (Varma et al. 1994; Yao et al. 1993). The detection of mitochondrial rRNA with a biotin-based system (Escaig–Haye et al. 1991; Binder et al. 1986) therefore requires additional analysis by studies of endogenous biotin which is specially concentrated in mitochondriae, associated with the Krebs cycle enzymes.
Detection of hybridized digoxigenin-labeled probes is mediated by high-affinity anti-digoxigenin antibodies conjugated to either alkaline phosphatase, peroxidase, fluorescein and rhodamine or revealed by a secondary antibody bound to colloidal gold. The use of unconjugated anti-digoxigenin antibodies with conjugated secondary antibodies enhances the detected signal (Figure 3e). It is interesting to note that a two-step colloidal gold immunodetection of digoxigenin always appears better than a two-step immunodetection of biotin (compare Figures 5 and 6), a result in agreement with the observations of Li et al. (1993) in their histochemical and blotting detection of glycoconjugates. However, the reasons for this discrepancy remain unclear, and it appears that the efficiency of the probe labeling might be better with the use of digoxigenin instead of biotin, whatever the technique used (nick translation or random priming). Actually, it was observed that as little as 10 pg of digoxigeninated albumin could be visualized in Western blot, whereas the limit of visualization for the biotinylated product was 500 pg (Brunet et al. 1994).
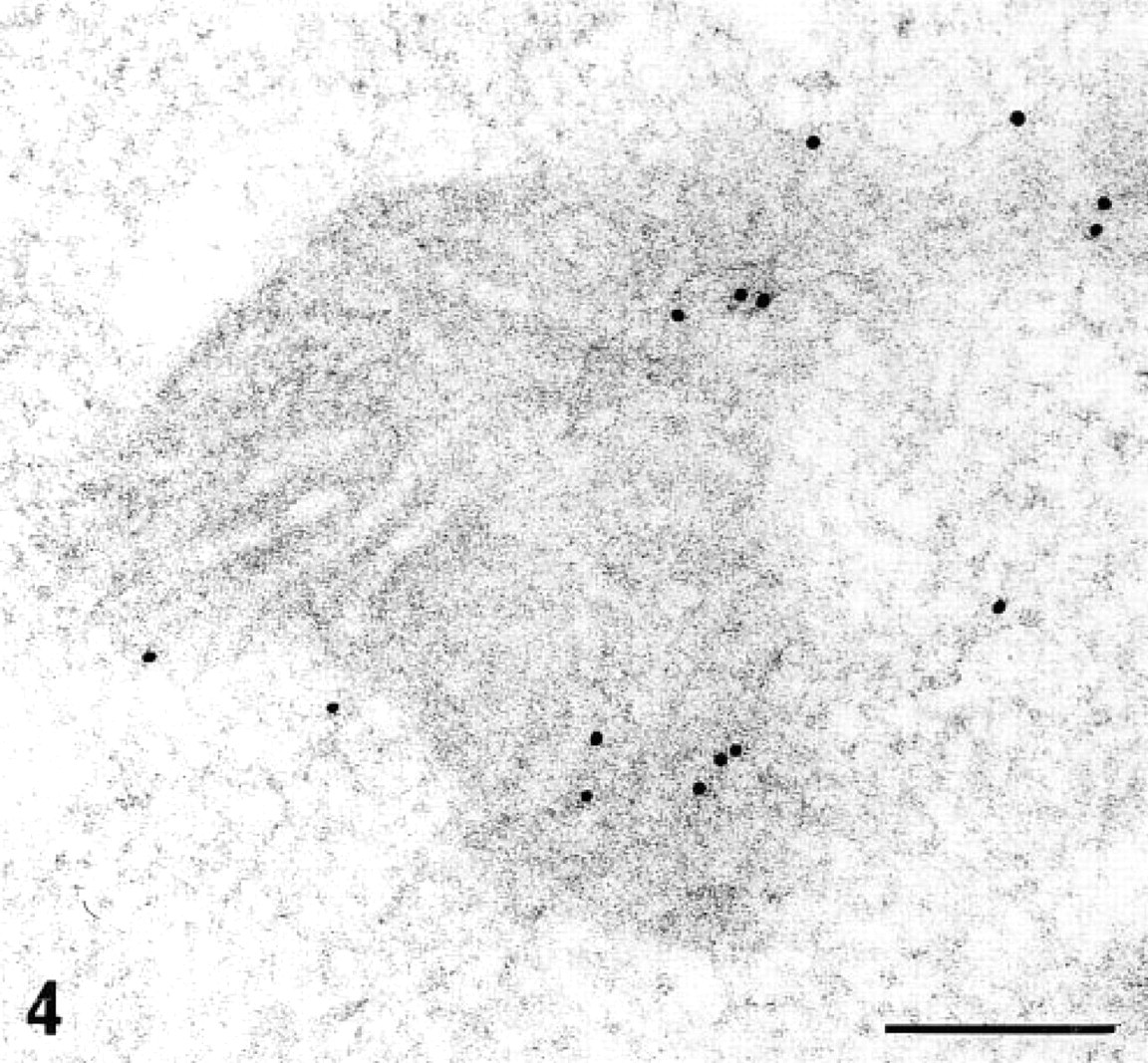
EM-ISH with the biotinylated probe for α1(I)-collagen mRNA and two-step immunogold detection (monoclonal antibiotin antibody and goat anti-mouse 10-nm colloidal gold) on a kidney tubule cell (old obese Zucker rat). In the tubule cell, many gold particles were restricted to mitochondria. Bar = 0.2 μm.
Peroxidase-conjugated avidin/streptavidin or antibodies have been largely used in the detection of biotin- or digoxigenin-labeled hybridizing probes, both in LM-ISH and EM-ISH. However, signals obtained with the use of peroxidase systems must also be discussed, because endogenous peroxidase activity can sometimes interfere with the true labeling of the section. In our EM-ISH detection of Type I collagen mRNA in kidney sections, we observed that endoplasmic reticulum was faintly stained in control sections in the absence of probe. Alkaline phosphatase also can interfere with the true labeling and attention must focus on reducing the endogenous phosphatase by using levamisole (see comments in Panoskaltsis–Mortari and Bucy 1995).
In conclusion, biotin and digoxigenin have been extensively used in non radioactive ISH. The size of the label and the optimal amount of labeled nucleotides incorporated with the probe have been clearly defined for performance of high-resolution ISH at both the light and the electron microscopic level. In routine studies, LM-ISH is performed on cryosections of frozen material or on dewaxed, permeabilized sections of paraffin embedded tissues. Labeled probes and detecting systems enter these sections more or less easily. If the number of target nucleotide sequence copies is high and is well located in a given structure, all detection systems—avidin or streptavidin, antibodies, peroxidase markers—can be used, the noise given by endogenous biotin and/or peroxidase being far below the detected hybridized signal.
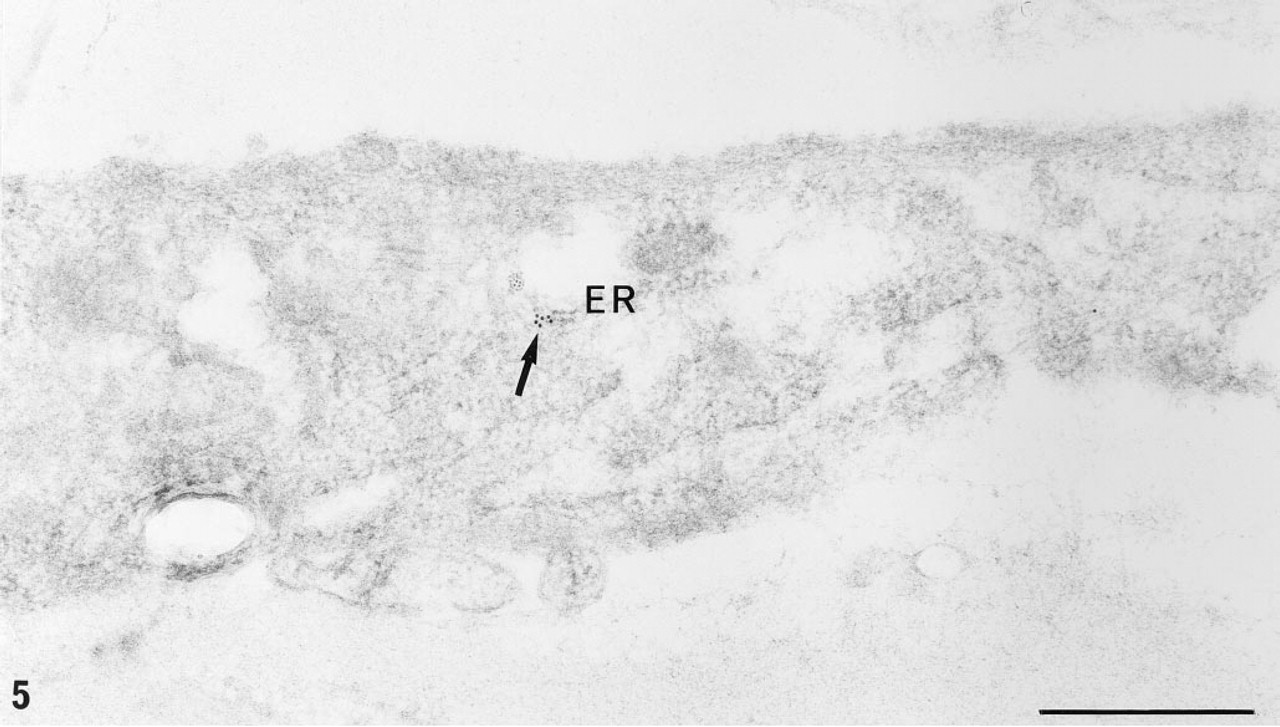
EM-ISH with the biotinylated probe for α1(∗∗∗I)-collagen mRNA and two-step immunogold detection (monoclonal anti-biotin antibody and goat anti-mouse 5-nm colloidal gold) on a fibroblast in interstitial fibrosis area (old obese Zucker rat). Sparse gold particles are seen on the fibroblast endoplasmic reticulum (arrow). ER, endoplasmic reticulum. Bar = 0.5 μm.
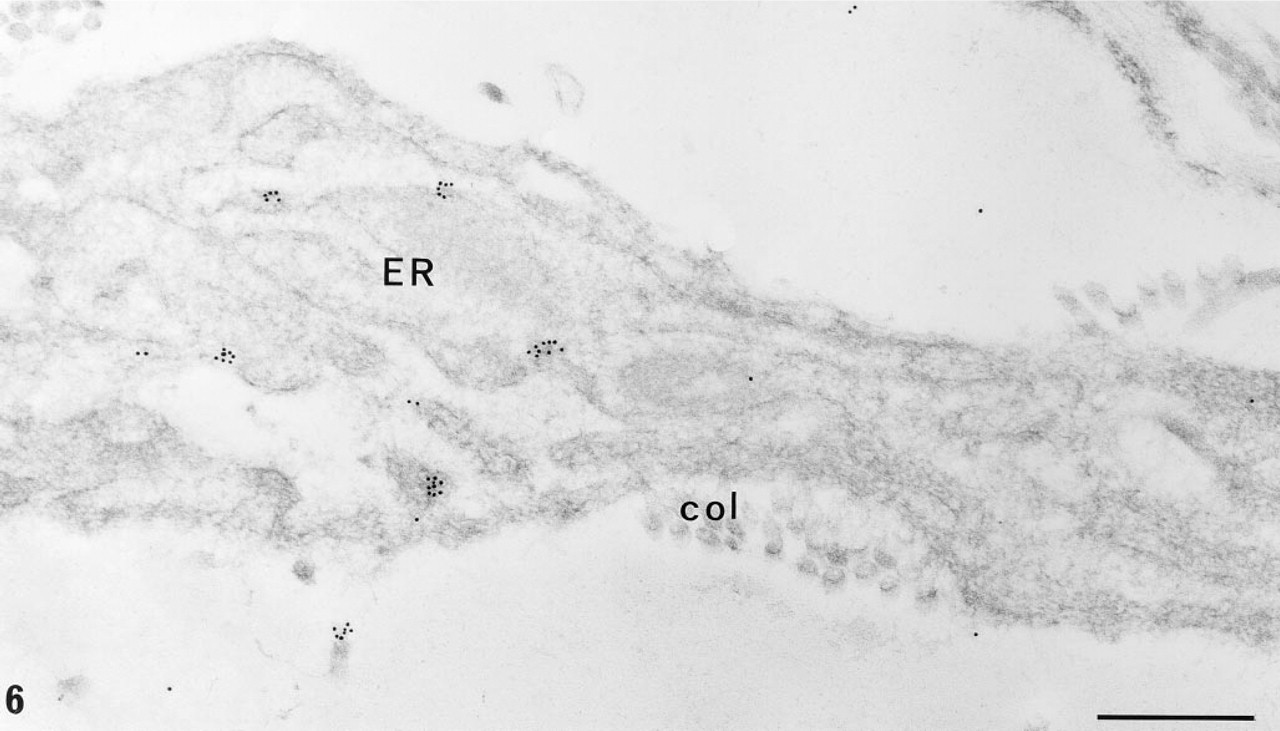
EM-ISH with the digoxigeninated probe for α1(∗∗∗I)-collagen mRNA and two-step immunogold detection (monoclonal anti-digoxigenin antibody and goat anti-mouse 10-nm colloidal gold) on a fibroblast in interstitial fibrosis area (old obese Zucker rat). Several clusters of gold particles are seen on the fibroblast endoplasmic reticulum (arrow). Digoxigenin-labeled probes afford a greater signal than biotinylated ones on the same material. ER, endoplasmic reticulum; col, collagen fibers. Bar = 0.5 μm.
In other situations, e.g., LM-ISH on semithin sections or EM-ISH in post-embedding techniques, the hybridization of labeled probes with the target nucleotides is limited by the ability of the probe to enter the section and by the accessibility and orientation of the target at the surface of the sections (Figures 3a and 3b). To overcome this difficulty, some investigators have developed an etching procedure for the sections, but the cell structure is so damaged that this procedure can be used only when nuclear targets are considered. In our experience, pretreatment was detrimental to detection of any mRNA in the extranuclear domain and induced non-specific labeling of the section. Regarding the probe and the detection system, when the number of copies is high and the target nucleotides well confined to a given structure, the use of biotin and or peroxidase can again be considered. However, we estimate that for all postembedding ISH studies, digoxigenin is a much more efficient label because endogenous biotin interference is avoided. To detect the digoxigenin moities, the use of antibodies conjugated to either fluorescent dyes or colloidal gold will avoid the potential noise caused by endoperoxidase or phosphatase. Colloidal gold signal can be enhanced with silver and could therefore be a useful conjugate, even for LM-ISH.
Guidelines for EM-ISH of mRNA
Because only small amounts of mRNA are exposed at the surface of embedded tissue sections with their longitudinal axes parallel to it, the hybridization signal in EM-ISH is inevitably weak. Consequently, careful technical attention must be paid, as the key points in postembedding EM-ISH are discrimination between specific and nonspecific labeling and hybridization efficiency.
Tissue Preparation
In our EM-ISH studies in kidney tissues, we found that the best morphological preservation is achieved when kidney samples are fixed for 4 hr in freshly prepared 4% paraformadehyde/0.1 M sodium cacodylate solution, pH 7.4. A 2-hr fixation step yields poor cytoplasmic morphology, with disrupted organelles and loss of mitochondrial cristae. Longer periods of fixation, or addition of glutaraldehyde to the fixative solution, although improving structural preservation, diminish hybridization efficiency. Tissue must be embedded in hydrophilic medium [such as Lowicryl K4M (TAAB; Reading, UK)], which polymerizes at low temperature (≤—20C). Embedding in other media that polymerize at room temperature or at 50–60C reduces the hybridation signal. The use of alcoholic stains, ethanol–uranyl acetate (EUA) and ethanol–phosphotungstic acid (EPTA), as described by Horowitz and Woodcock (1992), significantly improves the contrast of Lowicryl sections without any “pepper effect,” contamination by small, dense and coarse deposits. This enables intracytoplasmic structures and cell types to be precisely identified. EUA and EPTA solutions are prepared by mixing a 2% aqueous solution of uranyl acetate or phosphotungstic acid, respectively, with an equal volume of absolute ethanol immediately before use. Sections are doubly stained by floating the grids on a drop of EUA for 4 min and then on a drop of EPTA for 2 min, each step being followed by a washing step (jet of distilled water) for 30 sec.
Probe Preparation
As discussed in this review, digoxigenin must be preferred to biotin. Labeling of probes can be achieved either by nick-translation or by random priming, although Boehringer (Mannheim, Germany) manufactures a digoxigenin labeling kit based only on the random priming technique.
Labeling efficiency is routinely estimated by dot-blotting on membranes, as follows. Serially diluted probe DNA is spotted onto a nylon membrane. After fixation of DNA by brief exposure to UV radiation and pre-incubation with a blocking solution (Boehringer), the membrane is incubated with anti-digoxigenin antibody conjugated to alkaline phosphatase (Boehringer) for 30 min, then reacted with substrate solution prepared from a tablet of 5-bromo-4-chloro-3-indolylphosphate/nitroblue tetrazolium (BCIP/NBT: Sigma, St Louis, MO) for 15 min in the dark. Incorporation of digoxigenin is estimated from the intensity of the blue-purple dots, with reference to dots of standard labeled DNA (see Figure 2 as an example of digoxigenin- or biotin-labeled DNA).
Hybridization Procedure
Extremely varied conditions for hybridization (temperature and duration) have been proposed. In our hands, the most efficient ISH is obtained with a hybridization period of 16 hr (the first hour at 60C and the subsequent 15 hr at 45C). We found that it is essential to use and to maintain tiny drops (2 μl) of hybridization solution at a constant volume and original reagent concentration during the procedure. This is done by humidifying the hybridization chamber with the hybridization mixture minus the probe. At such a high temperature and long hybridization time, the small drops used are susceptible to dilution (if a standard buffer or plain distilled water is used to humidify the chamber) or to evaporation (if humidity levels are inadequate). Both situations would drastically change the concentration and efficiency of the probe. Pretreatment of sections with etching reagents and digesting enzymes must be avoided. The ISH mixture contains 50% deionized formamide, 10% dextran sulfate, 2 × standard saline citrate (SSC), and 10 μg/ml salmon sperm DNA. The labeled probe is added to the ISH mixture at a final concentration of 5–10 ng/μl. It is then denatured in boiling water and immediately transferred to ice before use. For hybridization, the grids are floated on drops (2 μl) of hybridization solution placed on Parafilm (Amercian National Can; Greenwich, CT) stuck to the bottom of a Petri dish. The dish is placed in a moist chamber containing two facial tissues (Lotus; Louviers, France) onto which 15 ml of ISH mixture, minus the probe, is poured. Hybridization is run for 1 hour at 60C and 15 hr at 45C. A brief post-ISH wash is done at room temperature by floating the grids on distilled water (1 min) and then on buffer T (Tris-HCl, pH 7.5, containing 0.1% bovine serum albumin, 0.1% fish gelatin, and 0.05% Tween) (twice for 3 min).
Probe Detection
Digoxigenin-labeled probes are commonly detected by a two-step immunogold system. The grids are incubated for 1 hr at room temperature with a mouse monoclonal anti-digoxigenin antibody (Boehringer) diluted 1:3000 in Tris buffer saline (TBS) containing normal rat serum (same volume as the anti-digoxigenin antibody) and then, after rinsing in buffer T (twice for 5 min) with a goat anti-mouse antibody conjugated to 10-nm gold particles (GAM-G10: Amersham Life Science, Little Chalfont, UK) (diluted 1:10 in TBS) for 1 hr.
Controls
Controls for in situ hybridization and the detection procedures are as follows: (a) ISH without the probe, followed by identical detection steps; (b) ISH followed by incubation with the second antibody conjugated to 10-nm gold (GAM-G10), omitting the incubation step with anti-digoxigenin antibody; and (c) treatment with RNAse (50 μg/ml) for 1 hr at 37C before ISH.
Footnotes
Acknowledgements
We thank Dr Srinivas Kaveri for helpful discussions during the preparation of the manuscript and Mr. Michel Paing for photographs.