
Abstract
We used the ELF-97 (
Keywords
NUCLEIC ACID HYBRIDIZATION with radioactively labeled probes has been used for several decades to detect complementary nucleic acid targets present on chromosomes, in cells and tissues, and bound to filter membranes. However, the use of probes labeled with radioisotopes has drawbacks, including the short half-life of some isotopes, rapid radiolysis of 32P-labeled probes, lack of spatial resolution of signals, and hazards associated with radioactivity. In addition, disposal of radioactive waste has become a serious problem. To overcome these drawbacks, a number of direct and indirect non-radioactive methods for labeling hybridization probes have been devised. Direct methods involve the covalent attachment of a detectable label to the nucleic acid probe, by chemical means (Kessler 1992) or by enzymatic means (Dirks et al. 1991; Wiegant et al. 1991). Indirect labeling employs a hapten, such as biotin (Gebeyehu et al. 1987; Chollet and Kawashima 1985; Langer et al. 1981), digoxigenin (Höltke et al. 1992; Kessler 1990,1991), or fluorescein (Durrant et al. 1995), covalently attached to the nucleic acid probe, which is then detected using a specific binding protein, such as anti-biotin, avidin or streptavidin, anti-digoxigenin, or anti-fluorescein. The protein recognizing the hapten is itself conjugated to a fluorophore or to an enzyme, such as alkaline phosphatase or horseradish peroxidase. Alkaline phosphatase has been most commonly used in this application, either with a hapten linkage (Leary et al. 1983; Langer et al. 1981) or a direct covalent linkage (Jablonski et al. 1986; Renz and Kurz 1984) to the hybridization probe.
These enzymes are most commonly used in combination with chemiluminescent or chromogenic substrates. The most widely used chemiluminescent substrate, used most frequently with alkaline phosphatase, is adamantyl 1,2-dioxetane aryl phosphate (AMPPD), which emits light at 477 nm on decomposition (Bronstein et al. 1990). This substrate is useful for detecting nucleic acids on filter membranes. However, because the reaction product rapidly diffuses from the site of enzymatic activity, it is not used for in situ hybridization, which requires high spatial resolution of signals. One of the most popular chromogenic methods for detecting alkaline phosphatase is based on the formation and precipitation of two highly insoluble products by sequential dephosphorylation of 5-bromo-4-chloro-3-indoyl phosphate (BCIP) and oxidation with nitroblue tetrazolium (NBT) (McGadey 1970). An alternative substrate is 3-hydroxy-2-naphthoic acid 2,4-dimethylanilide (naphthol AS-MX) which in combination with Fast Red TR, produces a red fluorescent precipitate (Speel et al. 1992).
The chromogenic substrates NBT and BCIP have been used to detect highly repetitive DNA regions on chromosomes (Weier et al. 1991), mRNA in cells and tissues (Kricka 1992), and DNA immobilized on membranes (Leary et al. 1983), but with low sensitivity in the latter application. Naphthol AS-MX has been localized to a DNA sequence in metaphase spreads and interphase nuclei (Speel et al. 1992) but not to mRNA in cells or tissues or to nucleic acids bound on membranes. The alkaline phosphatase substrates HNPP (2-hydroxy-3-naphthoic acid-2'-phenyl-anilide phosphate) and HBNP (3-hydroxy-N-2'-bi-phenyl-2-naphthalenecarboxamide phosphate ester) have been used to detect DNA hybridization on filters (Kagiyama et al. 1992,1995; Fujita et al. 1994). HNPP has also been used in combination with Fast Red TR to detect specific bacterial cells with rRNA-targeted oligonucleotide probes (Yamaguchi et al. 1996) and to detect targets on chromosomes, but only after a 2-hr incubation with the azo dye-coupled substrate (Kagiyama et al. 1995). Biotinylated or fluorophore-labeled tyramides have been successfully used as substrates for horseradish peroxidase to detect genes on chromosome spreads (Kerstens et al. 1995) but have not been used for in situ detection of mRNA or for detecting DNA hybridization on filter membranes. Although these substrates possess several advantages, such as signal amplification and long-term hybridization probe stability, they are still not as sensitive as radioisotopes, possibly because of low signal-to-noise ratios.
We have developed a novel substrate for alkaline phosphatase which belongs to a class of substrates that yield intensely fluorescent, photostable precipitates on enzymatic cleavage (Haugland et al. 1994, 1995; Haugland 1993). The fluorescent signal can easily be distinguished from most cell or tissue autofluorescence (Larison et al. 1995) and pigmentation because of its spectral properties, and therefore can be used in combination with a wide variety of counterstains and other fluorophores (Larison et al. 1995; Paragas et al., submitted for publication), including fluorescein (Brumback and Wade 1996). We refer to these compounds as the ELF-97 (
Materials and Methods
Reagents
Difco Chromosome medium, Dulbecco's minimum essential medium (D-MEM), bovine calf serum,
Cell Culture
CRE BAG 2 (NIH 3T3 mouse fibroblasts, transfected with Moloney murine leukemia virus bearing the E. coli lacZ gene) and MDCK (Madin-Darby canine kidney) cells were purchased from American Type Culture Collection (Rockville, MD). Cells were grown to near confluency in D-MEM buffered with sodium bicarbonate and 10 mM N-[2-hydroxy-ethyl]piperazine-N'-[2-ethanesulfonic acid] (HEPES) buffer and supplemented with 10% bovine calf serum, 2 mM
mRNA In Situ Hybridization
Cells. CRE BAG 2 cells were fixed with 3.7% formaldehyde in PBS (145 mM NaCl in 150 mM phosphate buffer, pH 7.2) for 15 min, then rinsed twice in 2 X saline sodium citrate (SSC = 300 mM sodium chloride, 30 mM sodium citrate, pH 7.0) for 1 min each time. Samples were then acetylated with 0.25% acetic anhydride in 0.1 M triethanolamine for 10 min, washed in 1 X SSC for 1 min, and in 10 X PBS for 10 min. Samples were next incubated in 0.2 M 2-amino-2-(hydroxymethyl)-1,3-propanediol (Tris)/0.1 M glycine, pH 7.4, and rinsed twice in 2 X SSC. Samples were dehydrated sequentially by washing briefly with 70% ethanol twice, 1 min each time, and once with 95% ethanol for 2–3 min, air-dried, prehybridized for 1 hr, and hybridized overnight at 42C. Prehybridization was in 25% formamide, 6 X SSC, 5 X Denhardt's solution (Denhardt's solution = 0.1% Ficoll 400, 0.1% polyvinylpyrrolidone, 0.1% bovine serum albumin) and 500 μg/ml herring sperm DNA; hybridization was in the same solution with 2 μg/ml biotinylated probes for actin or lacZ mRNA. The actin probe sequence (Hanukoglu et al. 1983) was CACGGAGTACTTGCGCTCAG-GAGGAGC and the lacZ probe sequence was TGTAAAAC-GACGGCCAGT. The following day, samples were washed with 2 X SSC three times at room temperature (RT), followed by three 0.1 X SSC washes, 5 min each time. For ELF detection, samples were blocked with ELF-97 mRNA Blocking Buffer for 1 hr and incubated with 10 μg/ml streptavidin-alkaline phosphatase in the same blocking buffer for 30 min. Samples were then washed with ELF-97 mRNA Wash Buffer three times, 5 min each time, and equilibrated in ELF-97 mRNA Developing Buffer for 5 min. Samples were then incubated with a 1:10 dilution of the ELF-97 phosphatase substrate and a 1:1000 dilution of substrate additives 1 and 2. For fluorescein detection, samples were blocked with ELF-97 mRNA Blocking Buffer for 1 hr and incubated with 10 μg/ml of fluorescein-streptavidin in the same blocking buffer for 30 min. Samples were washed in PBS with 0.1% Tween 20 several times, followed by a quick wash with 0.1 M Tris, pH 9. Photobleaching was performed as described (Paragas et al., submitted for publication). A comparison of relative photostability was made by determining the ratio of times at which the signal was 50% bleached for samples labeled with the fluoresceinated probe or the ELF substrate.
Tissue Sections. DNA fragments corresponding to the constant region of the mouse T-cell receptor β-chain (Cβ) were labeled with biotin-dUTP by nick-translation (Sambrook et al. 1989). Labeled probe was purified by passage through a QuickSpin G50 column.
Cross-sections of mouse lymph nodes embedded in paraffin (generous gifts from the laboratory of Dr. C. Garrison Fathman, Stanford University Medical Center, Stanford, CA) were dewaxed by extraction with xylene three times, 10 min each time. Slides were hydrated sequentially in 100%, 95%, 80%, 70%, 60% ethanol and water until the sections were clear. Tissues were then permeabilized with 0.2 M HCl for 20 min at RT, followed by 0.3% Triton X-100 in PBS for 15 min at RT and 17 μg/ml proteinase K in 0.1 M Tris, pH 7.5, and 50 mM ethylenediaminetetraacetic acid (EDTA) for 30 min at 37C. Tissues were rinsed in 0.2% glycine, pH 7.5, at RT and postfixed with 4% paraformaldehyde. To decrease nonspecific background, sections were acetylated with 0.25% acetic anhydride in 0.1 M triethanolamine for 10 min. Tissues were prehybridized in 50% deionized formamide, 2 X SSC, 10% dextran sulfate, 250 μg/ml herring sperm DNA, and 0.5% sodium dodecyl sulfate (SDS) for 2 hr at 37C and hybridized in the same solution with ∼2 ng/ml of biotinylated Cβ probe. The following day, slides were immersed in 4 X SSC and coverslips removed. Tissues were washed twice at RT in 2 X SSC for 15 min each time, followed by a quick rinse with ELF mRNA Wash Buffer. Blocking was done with ELF-97 mRNA Blocking Buffer for 30 min, followed by incubation with 10 μg/ml streptavidin-alkaline phosphatase in the same blocking buffer for 15 min at RT. Samples were washed with ELF-97 mRNA Wash Buffer, equilibrated in ELF-97 mRNA Developing Buffer for 5 min, and incubated with a 1:10 dilution of ELF-97 substrate with a 1:1000 dilution of substrate additives 1 and 2.
Embryos. Zebrafish embryos (24-hr stage for Gli and myoD, 16-hr stage for eng-3) were fixed in 4% paraformaldehyde in PBS (overnight at 4C for Gli and eng-3, 10 min at RT for myoD), dechorionated by hand, then dehydrated in methanol for 10 min at RT and 60 min at −20C. Embryos were then rehydrated by incubation in a graded series of methanol solutions in PBS: 5 min each in 75% methanol, 50% methanol, nd 25% methanol in PBS, and four changes of PBS with 0.1% Tween 20, 5 min each change. The embryos were then digested with 10 μg/ml of proteinase K in PBS with 0.1% Tween 20 for 20 min for myoD and Gli, or with 20 μg/ml proteinase K in the same buffer for eng-3. Embryos were then rinsed in 2% glycine in PBS twice for 30 min to stop the reaction for myoD and Gli, and in PBS with 0.1% Tween 20 for eng-3, twice for 5 min each time, and postfixed in 4% paraformaldehyde for 20 min.
In Situ Hybridization. Embryos were prehybridized at 65C for 2 hr for Gli, 42C for 1 hr for myoD, and 55C for 1 hr for eng-3, in a solution containing 50% formamide, 5 X SSC, 50 μg/ml heparin, 500 μg/ml tRNA, and 0.1% Tween 20. Probes were generated by in vitro transcription of Gli (a kind gift of John Postlethwait, University of Oregon, Eugene, OR), myoD, and eng-3 (kind gifts of Monte Westerfield, University of Oregon, Eugene, OR) cDNAs in the presence of digoxigenin-UTP, using the Genius 4 RNA Labeling Kit. Hybridization was at the temperatures indicated above, overnight in the same prehybridization solution with 100 ng/ml of Gli, myoD, or eng-3 probe. Posthybridization washes consisted of the following: washes at the hybridization temperatures indicated above with 75% hybridization solution and 25% 2 X SSC for 10 min, 50% hybridization solution and 50% 2 X SSC for 10 min, 25% hybridization solution and 75% 2 X SSC for 10 min, and 2 X SSC for 10 min; RT washes with 75% 0.2 X SSC and 25% PBS with 0.1% Tween 20, 50% 0.2 X SSC and 50% PBS, followed by 100% PBS with 0.1% Tween 20 for myoD and eng-3. One more wash with 25% 0.2 X SSC, 75% PBS was used for Gli. Detection. For labeling with the ELF substrate, samples were blocked with ELF mRNA Blocking Buffer containing 2% sheep serum for 1 hr at RT, incubated with preadsorbed anti-digoxigenin—alkaline phosphatase (0.15 U/ml for 2 hr for Gli, 1.5 U/ml for 1 hr for myoD and eng-3). For detecting myoD and eng-3 mRNA, samples were then washed several times with ELF mRNA Wash Buffer, equilibrated in ELF Developing Buffer for 5 min, and incubated with a 1:10 dilution of ELF substrate with a 1:1000 dilution of substrate additives 1 and 2. For detecting Gli mRNA using NBT/BCIP simultaneously with the ELF substrate and for labeling with NBT and BCIP alone, samples were blocked 1 hr in PBS containing 2 mg/ml BSA, 2% sheep serum, and 0.1% Tween 20, then incubated for 1–2 hr with a 1:5000 dilution of preadsorbed anti-digoxigenin-alkaline phosphatase conjugate in the same buffer. Preparations were then equilibrated for 5 min in 50 mM MgCl2, 100 mM Tris-HCl, pH 9.5, 100 mM NaCl, and incubated for 15–20 min in the same buffer containing 2.3 mg/ml NBT and 1.75 mg/ml BCIP or those concentrations of the colorimetric substrates plus a 1:10 dilution of the ELF substrate.
Chromosome In Situ Hybridization
Preparation of Metaphase Spreads. Lymphocytes from a normal adult woman were cultured for 72–96 hr in Difco Chromosome Medium at 37C. Cells were synchronized for 16–17 hr with 100 nM methotrexate and then incubated 5–5.5 hr with 0.01 mM thymidine. Mitosis was arrested and formation of spindle fibers interrupted with 0.2 μg/ml colce-mid for 15 min. Cells were pelleted gently and resuspended in a prewarmed (37C) hypotonic solution of 50 mM KCl for 20 min at 37 C. Cells were then fixed with methanol/acetic acid (3:1 v/v) for 20 min at RT and the fixative changed four or five times. Cells were dropped on slides cleaned with ethanol/HC (1:1 v/v) and further fixed on the slide with methanol/acetic acid, then dried with steam from a 70C waterbath for 30–45 sec. Slides were viewed with a phase-contrast microscope to check the quality of the metaphase spreads and to determine the mitotic index of the culture. Samples were stored at RT overnight before hybridization.
In Situ Hybridization. Slides were treated with 100 μg/ml RNAse A in 2 X SSC, under a plastic coverslip, for 1 hr at 37C, followed by four washes with 2 X SSC, 2 min each wash, and serial dehydration with 70%, 80%, and 95% ethanol. Chromosomes were denatured in 70% formamide/2 X SSC for 2 min at 70C, dehydrated in cold 80%, 90%, and 100% ethanol for 5 min each, and air-dried. The biotinylated centromeric DNA probe in hybridization buffer was denatured at 72C for 5 min and a 30-μl aliquot was applied to slides. Hybridization was allowed to proceed overnight at 37C. The following day, slides were washed with 50% formamide in 2 X SSC at 37C with agitation for 15 min, followed by an 8-min wash with 2 X SSC.
Detection. For labeling using the ELF-97 substrate, slides were first blocked in ELF mRNA Blocking Buffer for 1 hr, incubated with 10 μg/ml streptavidin-alkaline, phosphatase in the same solution for 30 min, washed several times with ELF-97 mRNA Wash Buffer, then equilibrated with ELF Developing Buffer. Samples were then incubated with a 1:20 dilution of the ELF-97 substrate and a 1:1000 dilution of substrate additives 1 and 2 in ELF Developing Buffer for 5 min. For labeling with fluorescein, slides were first blocked in PBS containing 3% BSA and 0.1% Tween 20 for 1 hr, incubated with 10 μg/ml fluorescein-avidin, followed by the same concentration of biotinylated anti-avidin and then fluorescein-avidin. In some cases, a second round of amplification was required to obtain sufficient signal.
Filter Hybridization
Fivefold dilutions of M13mp18 RF DNA digested with BamHI and HindIII were electrophoresed on a 1% agarose gel. After denaturation and neutralization, DNA was transferred overnight, by capillary action, to nylon filter membrane (Sambrook et al. 1989). The following day the membrane was rinsed quickly with water, air-dried, and baked for 1 hr at 80C. Baked filters were prehybridized in 25% deionized formamide, 6 X SSC, 5 X Denhardt's solution, and 500 μ/ml herring sperm DNA in a 37C waterbath and hybridized overnight in the same solution with 32P-labeled lacZ probe or 200 ng/ml biotin-labeled lacZ probe. The following day the filters were washed with 2 X SSC, 0.1% SDS twice for 20 min at RT, followed by a more stringent wash with 0.2 X SSC, 0.1% SDS for 30 min at 37C. After the posthybridization washes, the filter hybridized to the 32P-labeled probe was exposed to Kodak XAR 5 film overnight at −70C, with an intensifying screen. The filter hybridized to the biotinylated probe was blocked for 1 hr in ELF-97 mRNA Blocking Buffer and incubated for 30 min at RT with 10 μg/ml streptavidin-alkaline phosphatase in the same buffer, washed several times with ELF-97 mRNA Wash Buffer, and equilibrated for 5 min in ELF-97 mRNA Developing Buffer. The filter was then incubated overnight with a 1:5 dilution of the ELF-97 substrate.
In-gel Detection of Biotin-labeled HindIII-digested λ-DNA
Twofold dilutions of HindIII-digested λDNA were electrophoresed on a 0.8% agarose gel. After denaturing and neutralization (Sambrook et al. 1989), the gel was dried down for 1.5 hr with a BioRad Model 583 Gel Dryer using the gradient cycle setting at 50C. This setting raises the temperature 1C each minute until the dryer reaches 50C. The dried gel was rehydrated in water, blocked in ELF mRNA Blocking Buffer overnight, and incubated for 48 hr with 10 μg/ml streptavidin-alkaline phosphatase in the same blocking buffer. The gel was washed 10 times with ELF-97 mRNA Wash Buffer, equilibrated for 5 min in ELF-97 Developing Buffer, and incubated overnight with a 1:5 dilution of ELF-97 substrate.
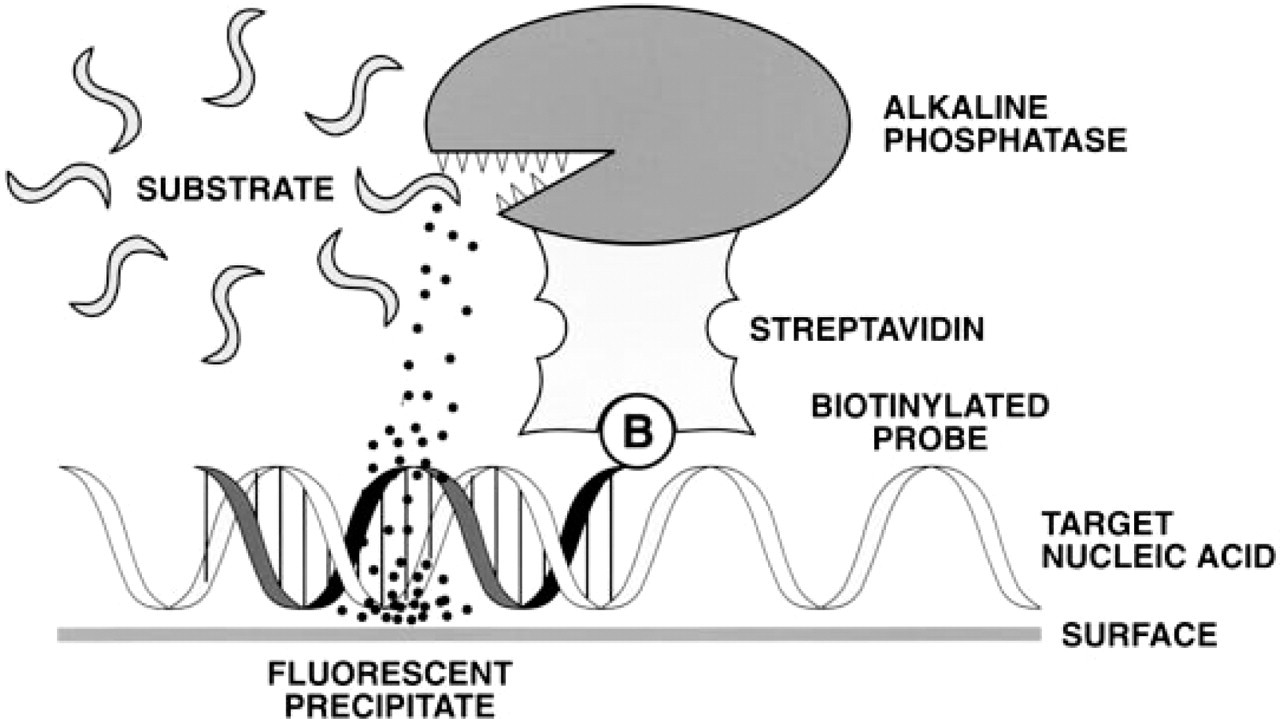
Schematic diagram showing the use of the ELF-97 phosphatase substrate in detection of nucleic acid hybridization.
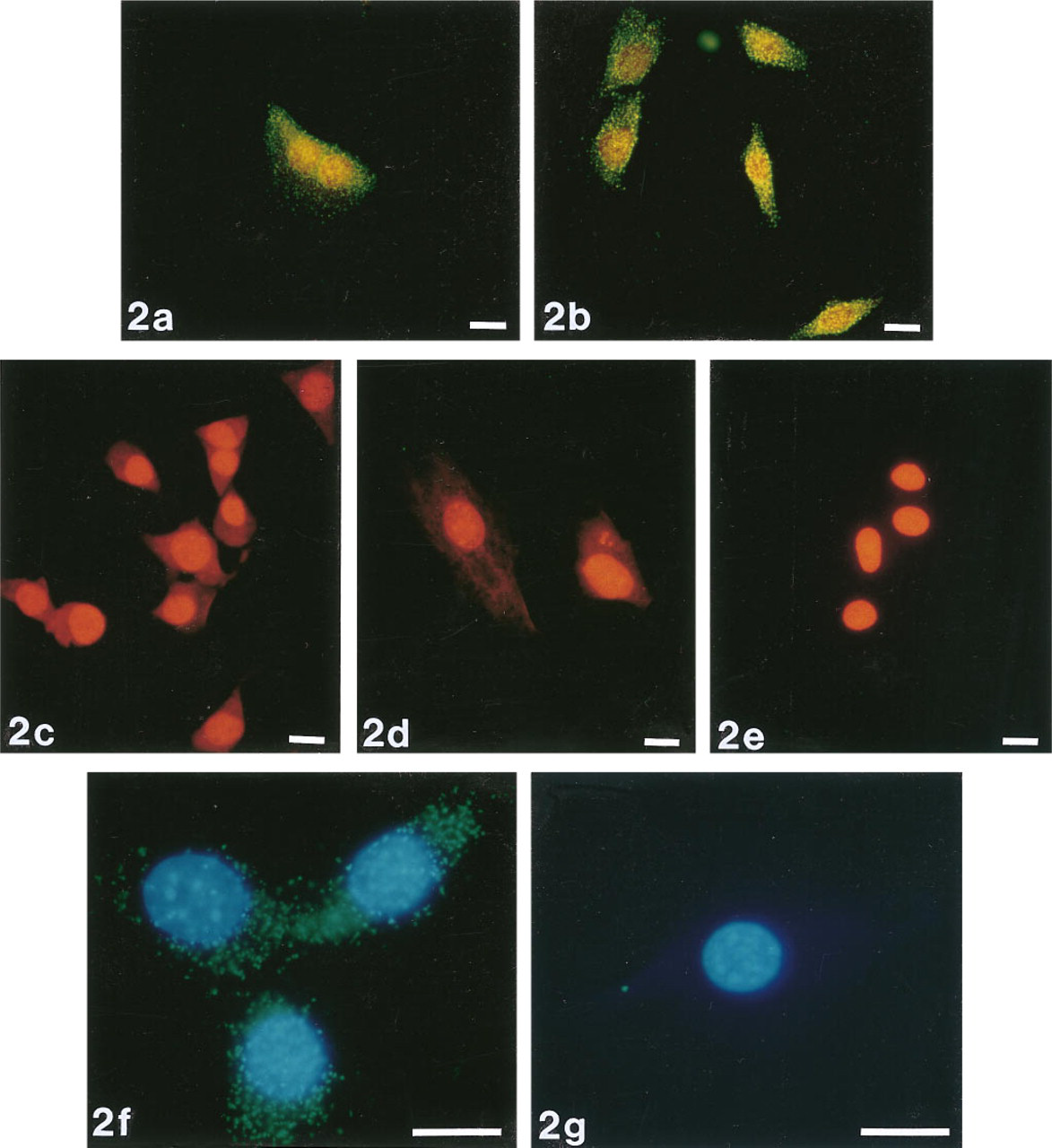
Detection of lacZ mRNA in cultured fibroblasts using the ELF-97 phosphatase substrate. CRE BAG 2 cells were probed with a biotinylated oligonucleotide directed against lacZ mRNA and then hybridization was detected with streptavidin-alkaline phosphatase and the ELF-97 phosphatase substrate. Panels show detection of lacZ expression (
Results
We previously demonstrated the successful use of the ELF-97 substrate in immunohistochemistry (Larison et al. 1995) and immunocytochemistry (Paragas et al., submitted for publication). In this report we show that these techniques can be adapted for fluorescence in situ hybridization, to detect specific mRNAs in cells and complex tissues and to detect DNA sequences on chromosomes or bound to filter membranes. A schematic depicting the basic methodology is shown in Figure 1.
The ELF-97 Substrate Can Be Used to Specifically Detect mRNA In Situ Hybridization in Cultured Cells
We used oligonucleotide probes labeled with a single biotin moiety to detect mRNA from a transfected gene (E. coli lacZ) and from a gene that is expressed abundantly in all organisms (actin). Signals were specific; positive and negative controls were easily distinguished from one another (Figure 2). In contrast, cells labeled with NBT and BCIP were indistinguishable from unlabeled cells (Figure 3). The ELF signals were 28-fold brighter and 32-fold more photostable than signals generated using oligonucleotide probes labeled with a single fluorescein moiety (Figure 4). ELF signals were visible using a X 40 microscope objective, whereas signals obtained using fluorescein-labeled probes were only visible with a X 100 oil-immersion lens. This was true both for samples labeled with the fluoresceinated probe and for those labeled with a biotinylated probe followed by fluorescein-streptavidin. Actin mRNA in MDCK cells, which appears in our experiments to be sequestered or highly localized, was also readily detected using the ELF substrate (Figure 5). Actin mRNA is known to be highly localized in mammalian fibroblasts (Alberts et al. 1994; Kislauskis and Singer 1992) and in differentiating myoblasts (Kislauskis et al. 1993).
The ELF-97 Substrate Can also Be Used to Detect mRNA Expression in Tissues
Anti-sense RNA probes labeled with digoxigenin-UTP were hybridized to myoD, eng-3, and Gli mRNA in zebrafish embryos and were detected using the ELF substrate and the colorimetric substrates NBT and BCIP (Figure 6). The ELF signal appears as yellow or yellow-green fluorescence against the green or blue yolk fluorescence. In the detection of Gli mRNA, the ELF signal co-localized with NBT/BCIP labeling (Figures 6a–6a); signals were not fully equivalent because a buffer optimal for NBT/BCIP labeling was used to label simultaneously with both substrates. The ELF-97 phosphatase substrate does not label well in that reaction buffer because the precipitate dissolves at high pH (Huang et al. 1992). However, it is clearly easier to detect the ELF signal than the signal from the colorimetric substrates against the dark tissue pigmentation. The yellow fluorescence was present for myoD mRNA on developing muscle cells along the embryonal axis (Figures 6g and 6h), as expected (Weinberg et al. 1996), but was absent from cells that were treated with RNAse A (Figure 6 i). Eng-3 mRNA detection showed similar patterns for the fluorescent (Figures 6j and 6k) and colorimetric substrates (Figure 6 l), with a primary region of labeling in the midbrain/hindbrain junction as expected (Ekker et al. 1992; Hatta et al. 1991). This labeled region appeared slightly larger in the sample labeled with the ELF substrate than in the sample labeled with the colorimetric substrate, perhaps indicating a difference in sensitivity for the two methods.
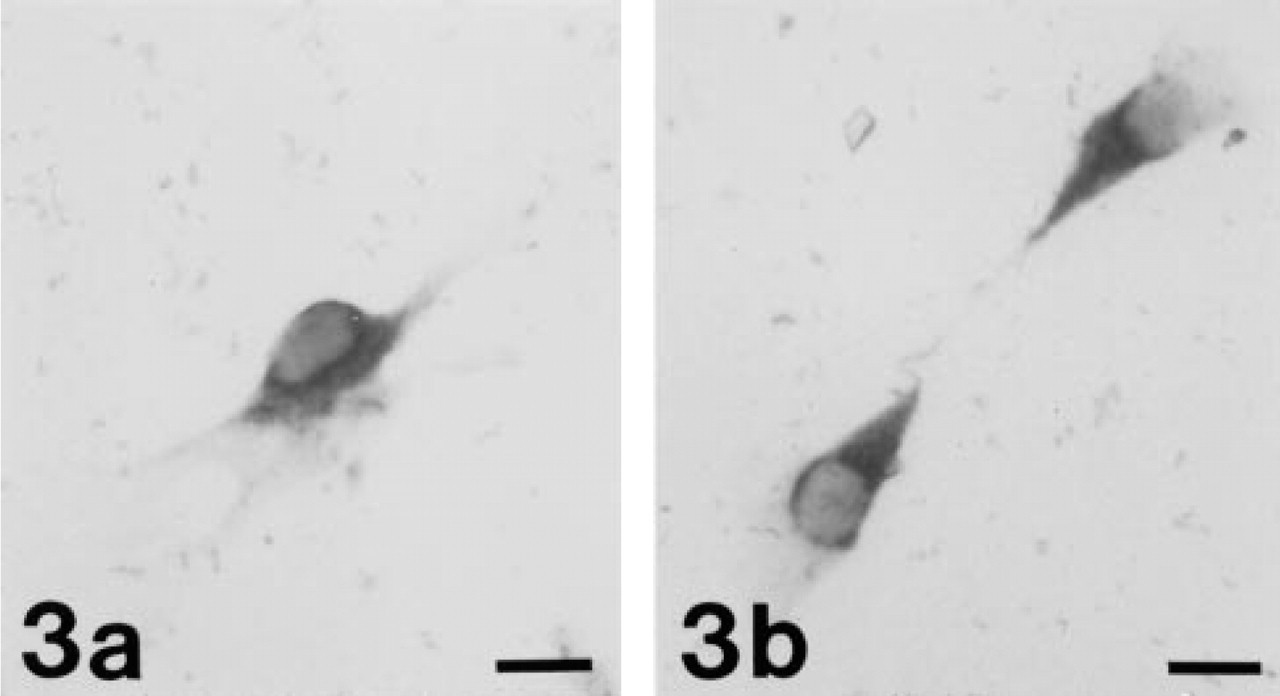
Use of NBT/BCIP to label biotinylated hybridization products. CRE BAG 2 cells were probed with a biotinylated oligonucleotide directed against the lacZ gene (
When anti-sense RNA probes corresponding to the constant region of the T-cell receptor β-chain were labeled with biotin-UTP and used in combination with the ELF-97 substrate in cross-sections of paraffin-embedded mouse lymph nodes, the ELF signal was localized on the T-lymphocytes only and was not seen on the cells lining the blood vessels (Figure 7). The ELF signal appeared as a bright green fluorescent precipitate against the red fluorescent propidium iodide counterstain, which identified the locations of the nuclei.
The ELF-97 Substrate Can Be Used to Detect Chromosome In Situ Hybridization
We localized the ELF signal to highly repeated alphoid DNA sequences (Figures 8a-8c), which are located at the centromeres of human chromosomes (Gray et al. 1985). ELF signals were visible even at very low magnifications. A X 10 microscope objective could be used to visualize the centromeric signals on interphase nuclei (data not shown), and a X 40 microscope objective for centromeric signals on metaphase spreads. On the other hand, the fluorescein signal could be resolved only using a X 100 oil-immersion lens (Figures 8d-8f). The signal-to-background ratio was also much better with the ELF-97 substrate than with fluorescein, and therefore the camera exposure time was much shorter.
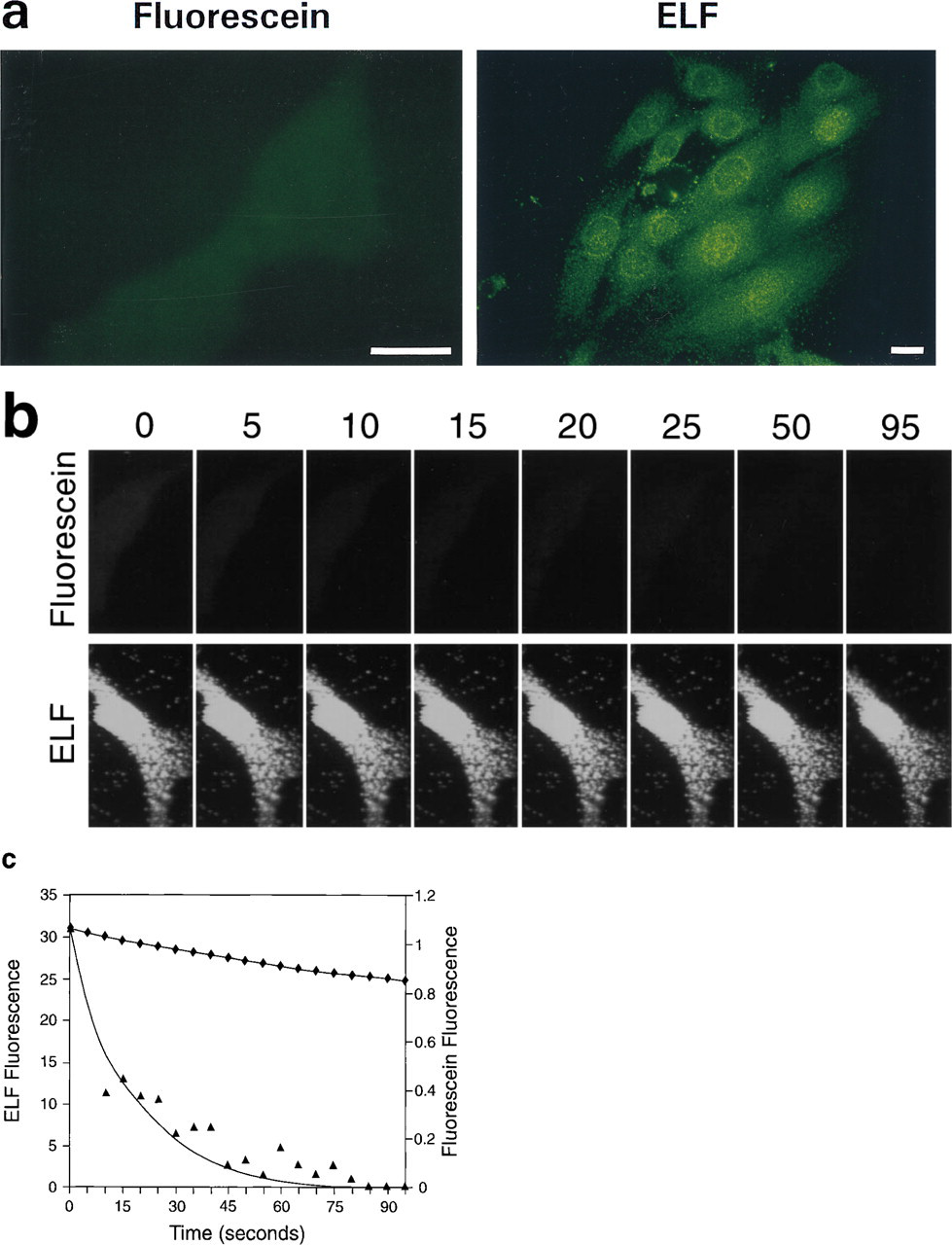
Comparison of the relative brightness and photostability of the ELF precipitate and fluorescein for detecting actin mRNA in fibroblasts. CRE BAG 2 cells were probed with oligonucleotides directed against actin mRNA, which were labeled with either a single biotin or a single fluorescein. Cells containing the biotinylated probes were then labeled using streptavidin-alkaline phosphatase and the ELF substrate. (
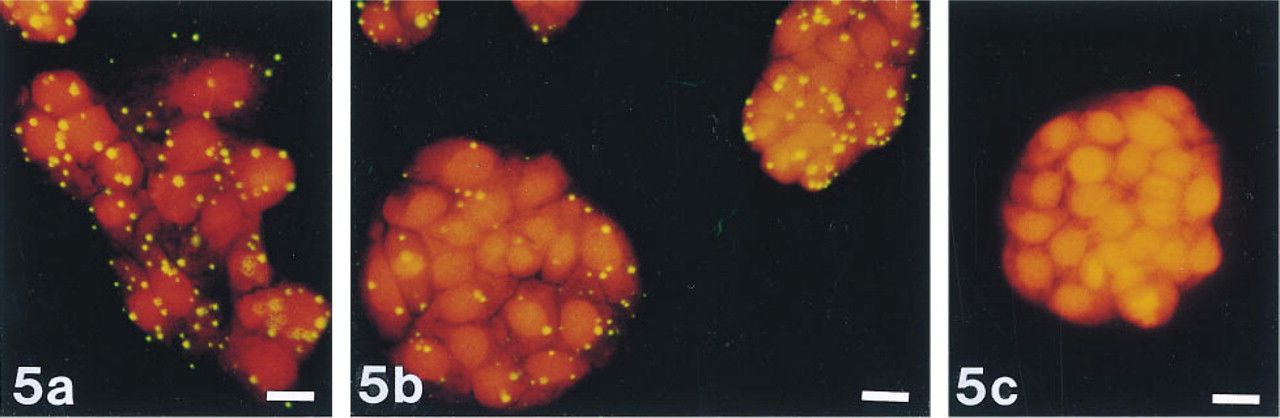
Detection of actin mRNA in MDCK cells using the ELF substrate. MDCK cells were probed with an oligonucleotide directed against actin mRNA, followed by incubation with streptavidin-alkaline phosphatase and the ELF-97 phosphatase substrate. Panels show detection of actin mRNA (
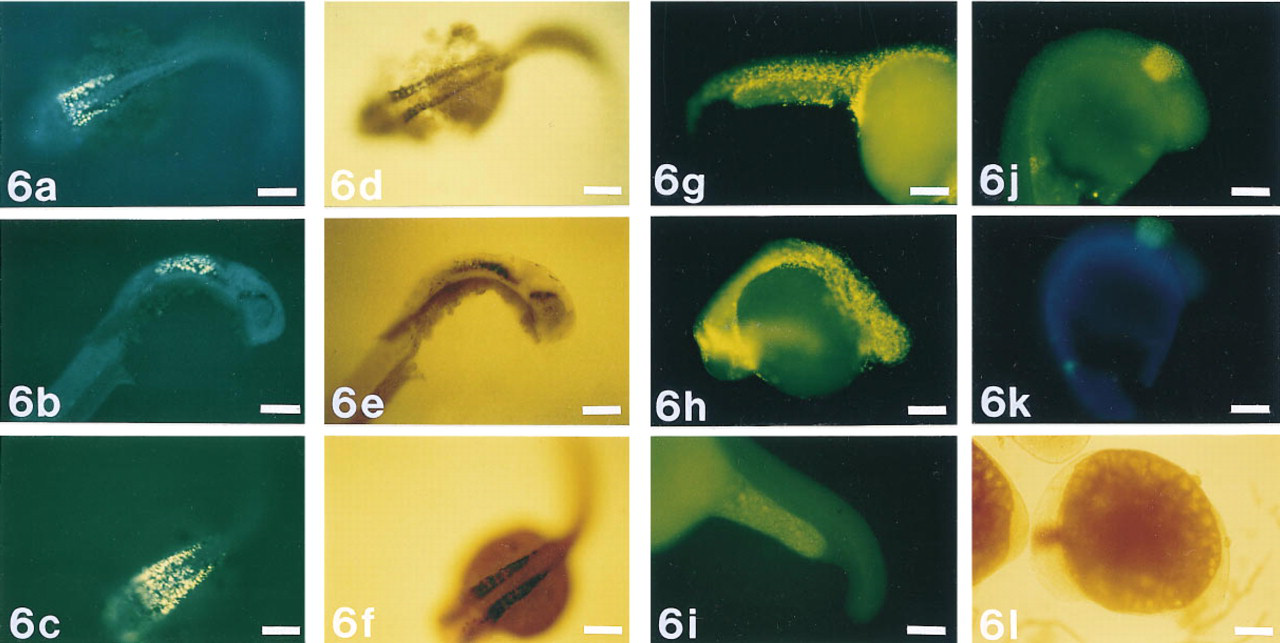
Detection of mRNA in situ hybridization to targets in zebrafish embryos. The ELF substrate and NBT/BCIP were used simultaneously to detect Gli mRNA (
The ELF-97 Substrate Can Be Used to Detect Hybridization Signals on Filter Membranes
Southern blots with HindIII and BamHI-digested M13mp18 RF DNA hybridized to either a 32P-labeled lacZ probe or a biotin-labeled lacZ probe, followed by streptavidin-alkaline phosphatase and the ELF substrate are shown in Figure 9. Digestion with these restriction enzymes generates two bands, a ∼6.3-
The ELF-97 Substrate Can Be Used for In-gel Detection of DNA
Biotinylated DNA was detected directly in an agarose gel using the ELF substrate (Figure 10 a). This suggests that the substrate can potentially be used to detect the products of in-gel hybridization. (Purrello and Balazs 1983; Tsao et al. 1983). The signal is relatively linear with respect to DNA content (Figure 10 b) and could be used to quantitatively assess the extent of hybridization in such samples.
Discussion
Fluorescence in situ hybridization (FISH) is a widely used method for localizing DNA sequences to specific chromosomal regions, for localizing specific RNAs to cells or regions of cells, and for measuring the relative level of gene expression in cells in culture or in complex tissues. Fluorescent labels with increased brightness and photostability would be desirable in all of these applications, particularly for detecting low copy number targets. Here we describe the use of the ELF-97 phosphatase substrate for amplifying hybridization signals. Because the substrate has been shown to be useful for immunohistochemistry (Larison et al. 1995) and immunocytochemistry (Paragas et al., submitted for publication) showing not only a high degree of signal resolution but also greatly increased brightness and photostability over conventional fluorophores in the latter study, we reasoned that it might yield the same advantages in the detection of chromosome and RNA in situ hybridization. We found that for RNA in situ hybridization these advantages were indeed conferred. Signals obtained form singly biotinylated probes with the ELF substrate were 30-fold brighter than those obtained using singly fluoresceinated probes. In addition, they were much more photostable. Signals developed in minutes, whereas those radioactive probes require days to weeks (Angerer and Angerer 1994). In addition, because of the large Stokes shift of the ELF precipitate, sample autofluorescence did not interfere and signals were easy to distinguish from pigmented cells and tissues. The fluorescence was easily detectable with standard fluorescence microscopy and photography with 35-mm film, and did not require sensitive CCD-based image acquisition and analysis systems. In addition, the photostability of the preparations enabled us to observe the sample repeatedly and to obtain multiple photographic exposures without significant sample bleaching.
However, for chromosome in situ hybridization, we found the usefulness of the substrate to be somewhat more limited. We show here the accurate localization of human centromeres on metaphase and interphase spreads, using probes directed against repetitive sequences. Such signals were extremely bright compared to fluorescein-labeled probes, even when several conventional amplification steps were employed to increase the fluorescein signals. However, when detection of single copy genes (p53) on metaphase chromosomes with cosmid probes was attempted, the ELF precipitate arose not only at the correct sites but also nonspecifically on other chromosomes. We suspect that this is probably due to an insufficiently high concentration of active alkaline phosphatase enzymes in the immediate vicinity of the target. Under such conditions, the enzymatic product accumulates slowly and perhaps does not reach the critical concentration required for precipitation before diffusion from the reaction site. In addition, there are probably fewer nucleation sites present on the target, so that microcrystals are able to diffuse away and precipitate on other regions of the sample. It is possible that the use of hybridization probes containing directly conjugated enzymes or the use of more heavily biotinylated or longer probes might alleviate this problem. It is also possible that cosmid probes can be used to detect targets in interphase nuclei, where accurate localization along a particular chromosome is not needed. The brightness and photostability of the signals on high copy number targets, coupled with the large Stokes shift of the dye, might still make it advantageous for chromosome FISH under some conditions. In particular, the ELF substrate can be used in combination with other fluorophores (Larison et al. 1995; Paragas et al., submitted for publication), including fluorescein (Brumback and Wade 1996), to provide a distinct color for fluorescent detection. In addition, because of its brightness it provides a useful tool for detecting signals on interphase nuclei, even at relatively low magnifications. This might be useful for applications in which large numbers of interphase nuclei must be assayed in a single microscope field, for general genetic characterization, or for detection of differences in the copy number of specific genetic targets (e.g., for ploidy analysis).
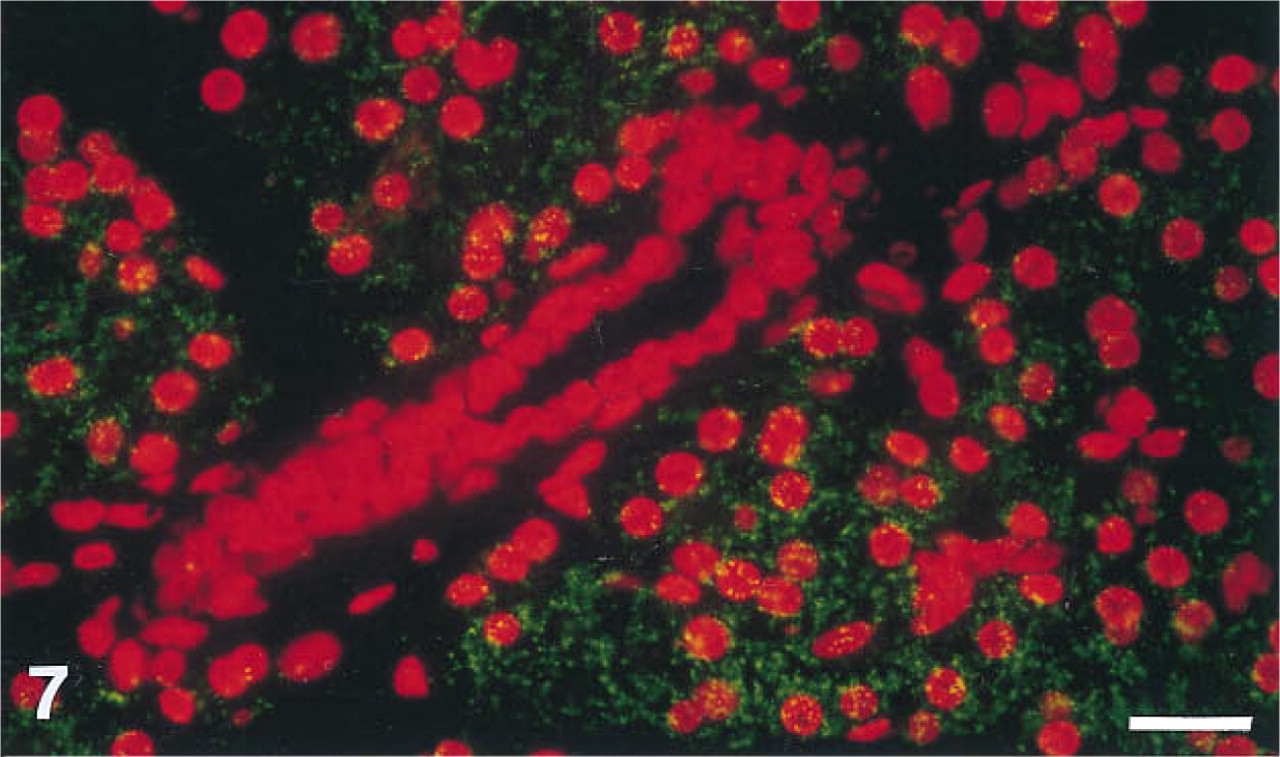
Detection of in situ hybridization to the T-cell receptor β-chain mRNA in mouse lymph node tissue sections. The ELF substrate gives rise to green fluorescence on T-cells but does not label the cells lining the blood vessel. The red fluorescence is due to propidium iodide, which indicates the location of all nuclei in the sample. The sample was photographed by double exposure through a rhodamine filter set and a UV excitation/fluorescein emission filter set with a Nikon Labophot microscope. Bar = 20 μm.
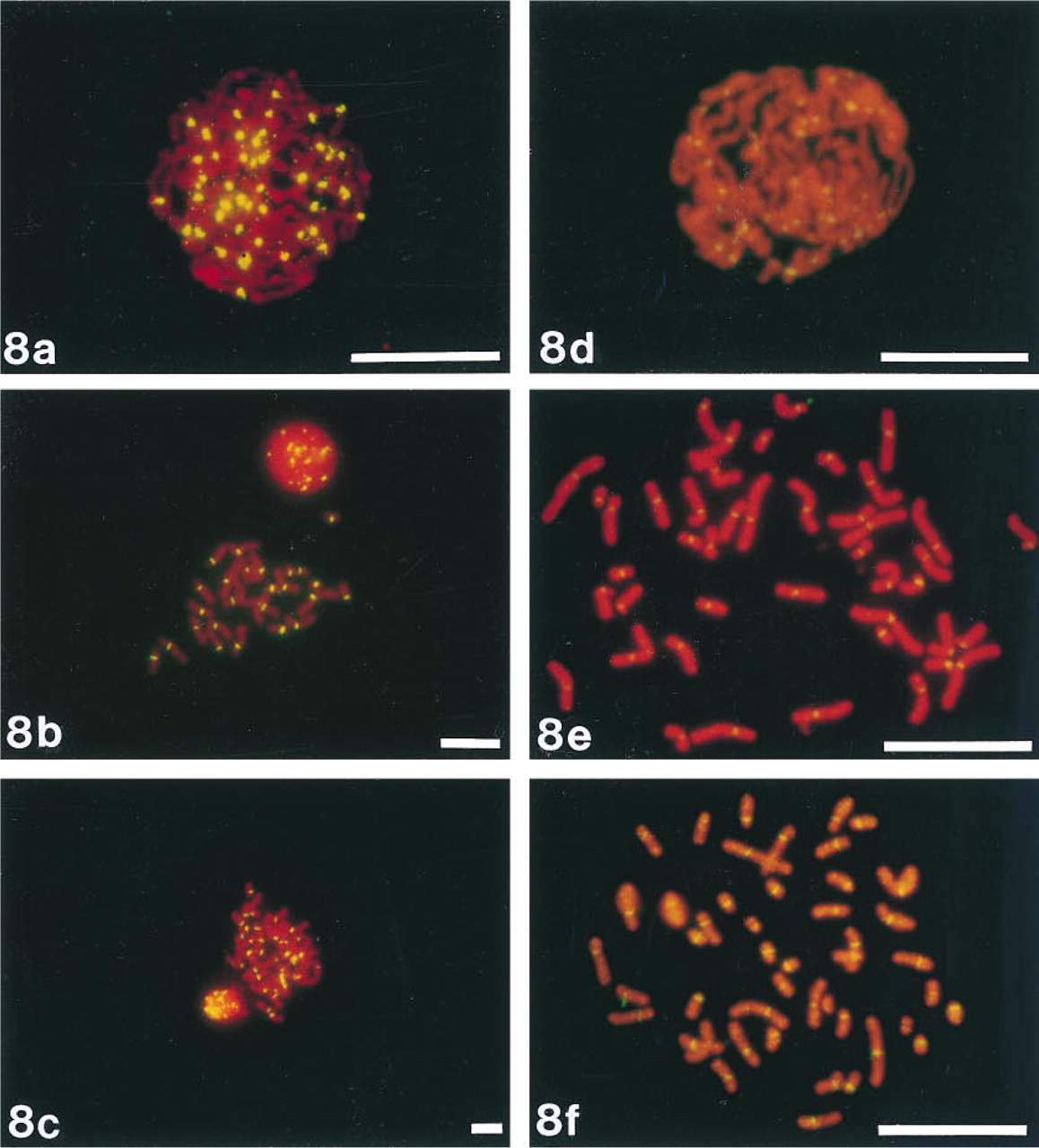
Comparison of fluorescein and the ELF-97 substrate for detecting in situ hybridization to human centromere alphoid repeat regions. Metaphase and interphase chromosome preparations were hybridized with a biotinylated probe directed against repetitive DNA in the centromere region. Samples were then incubated with streptavidin-alkaline phosphatase and the ELF substrate (
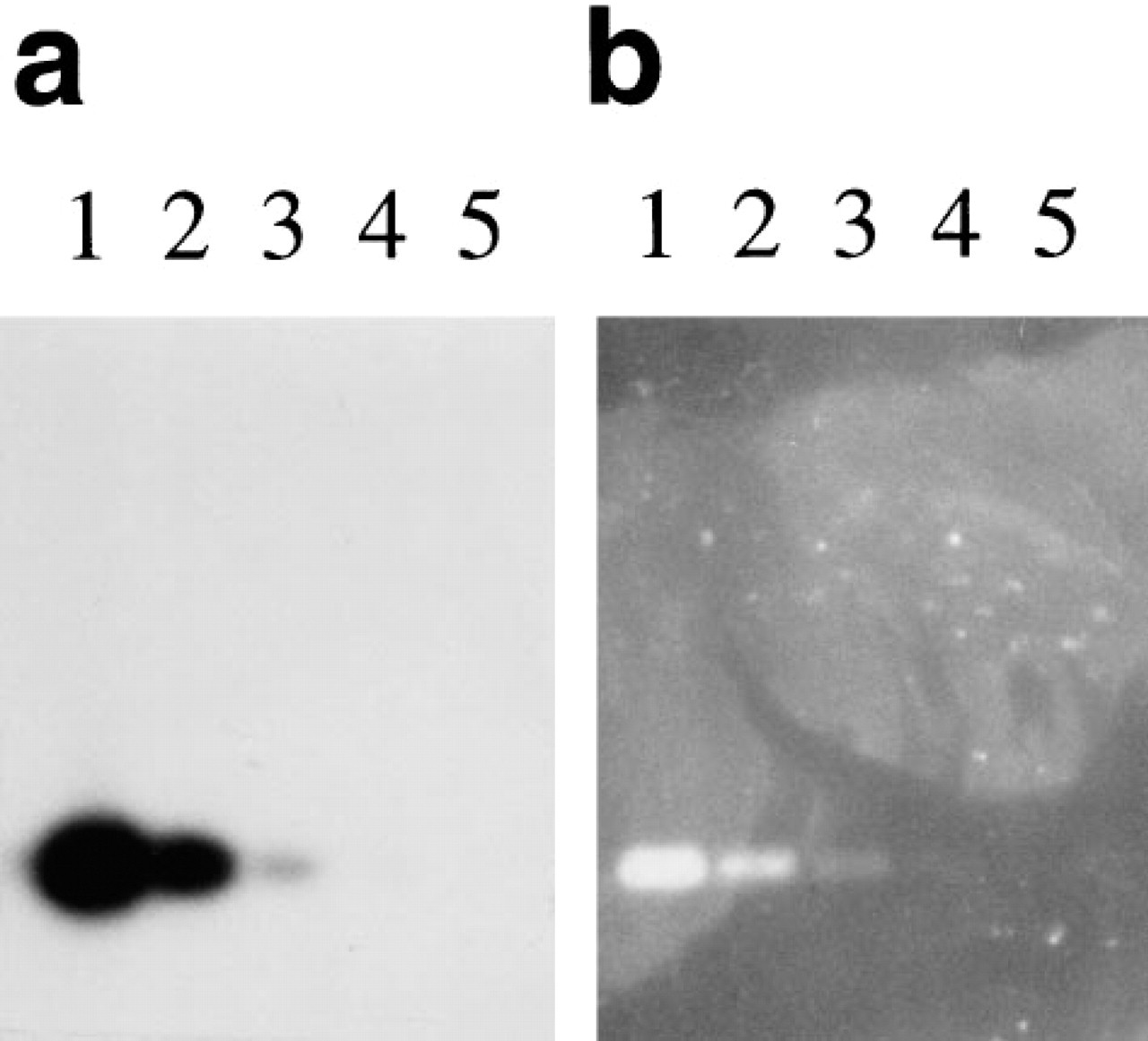
Southern blot comparing detection using the ELF substrate and radioactivity. Oligonucleotide probes with either a single biotin or a single 32P label at the 5' end were hybridized to a Southern blot containing BamHI/HindIII-digested M13mp18 RF DNA. The blot with the radioactive label was exposed to X-ray film in the presence of an intensifying screen (
We also show that the ELF substrate can be used to detect nucleic acid hybridization on filter membranes. For Southern blot detection, using singly labeled short oligonucleotide probes, the sensitivity was essentially the same as that achieved with radioactivity. The signal was extremely photostable, allowing quantitation several years after the blot was made, with little decrease in image quality. However, we found that when longer probes with multiple labels were used, although the sensitivity was still much better than that obtained with NBT/BCIP, radioactive probes provided greater sensitivity than the ELF-97 phosphatase substrate (data not shown). Our finding that the substrate can also be used to detect labeled DNA in gels indicates that it could be very useful for in-gel hybridization as well. Although this is not a methodology in common use, it avoids transfer to filters, which is not always an efficient process and can result in loss of sample and lack of uniformity across the blot.
In summary, the ELF-97 phosphatase substrate is particularly useful for detecting mRNA in situ hybridization, not only as a green fluorescent label with novel spectral properties but also to provide significant signal amplification and photostable preparations. For chromosome in situ hybridization it may be useful within a more limited sphere of applications, but is likely to be especially well-suited to clinical research, in the enumeration of signals on interphase nuclei. Although it can be used as a detection reagent for Southern analysis, it is unlikely to supplant AMPPD or other chemiluminescent substrates in that application, simply because for probes containing multiple labels it is not likely to have sufficient sensitivity. However, when used with single labeled synthetic oligonucleotide probes it is extremely sensitive. For microscopy, its greater than 100-nm Stokes shift makes it especially useful for working with autofluorescent samples and in combination with other fluorophores for either mRNA or chromosome FISH.
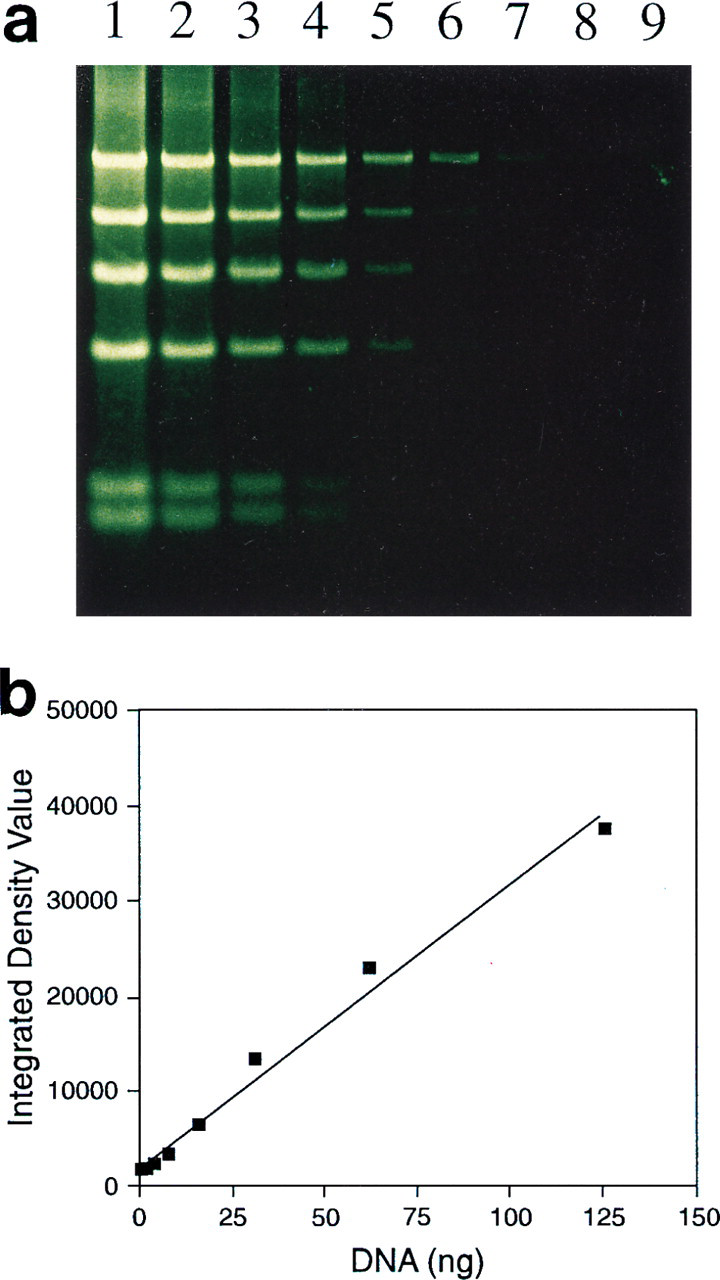
In-gel detection of biotinylated DNA. Biotinylated bacteriophage λ DNA digested with HindIII was separated on a 0.8% agarose gel, and the gel was partially dried and then incubated with streptavidin-alkaline phosphatase and the ELF-97 phosphatase substrate (
Footnotes
Acknowledgements
We gratefully acknowledge the help of Janell Bishop and Paul Millard in performing the photobleaching analysis and quantitative intensity comparisons, the preparation of DAPI-counterstained samples by Jennifer Kramer, the analysis of the stained gel by Deborah Smead of Alpha Innotech, the assistance of a number of Molecular Probes employees in editing the manuscript, and the contribution of Isamu Sato in the preparation of the figures. We are also grateful for the generosity of Monte Westerfield in allowing us to use his laboratory for zebrafish work, to Jeremy Wegner for help with zebrafish in situ hybridization, and for the generosity of Peter von Hippel for allowing us to use his laboratory space and reagents to perform experiments requiring radioactive labels. Mouse lymph node tissue sections were generously provided by Cariel Taylor-Edwards and Garry Fathman.