
Abstract
Two fibrillar collagens, the worm cuticular collagen and the vertebrate Type I fish scale collagen, both organized in a compact tissue, were localized by immunogold electron microscopy in resin sections after freeze-fixation and freeze-substitution. Identification of these two fibrillar collagens failed with the-use of postembedding labeling after conventional electron microscopic processing. Positive labeling of the Type I collagen was observed in sections of fish scales freeze-fixed by either slam-freezing or high-pressure freezing, freeze-substituted in acetone with or without osmium tetroxide, and embedded in LR White. The worm cuticular collagen was detected in sections of cuticle that were freeze-fixed, freeze-substituted (necessarily with osmium tetroxide added to acetone), and embedded in either LR White or Epon. It was also detected in specimens pre-fixed by aldehydes before freeze-fixation. The Type I fish scale collagen appears to be more sensitive than the fibrillar cuticular collagen of worms to the procedures employed for postembedding immunoelectron microcopy. Our results have shown that freeze-fixation and freeze-substitution preserved the antigenicity of the fibrillar collagens organized in a compact three-dimensional network, whereas immunolabeling failed after conventional electron microscopic procedures. These cryostabilization techniques appear to be of value to improve the immunolocalization of collagens.
Keywords
I
Conventional methods using aqueous fixations, dehydration, and embedding induce changes in the structure and composition of the tissues, including extraction, precipitation, displacement, and loss of antigenicity. To overcome the problems induced by chemical fixation and dehydration, alternative methods of tissue processing have been developed (Kellenberger and Hayat, 1991). Pre-embedding immunolabeling has provided invaluable data about the molecular topology of the collagen fibrils (Fleischmajer et al., 1990; Birk et al., 1988), but these immunoelectron microscopic techniques have been applied after disruptive treatments that may induce disorganization of the tissue architecture. Another strategy for concomitantly preserving ultrastructure and antigenicity of tissues involves cryotechniques (Nicolas, 1991; Nicolas et al. 1989), such as ultra-rapid freeze-fixation or high-pressure freezing followed by freeze-substitution, which improve the quality of the ultrastructural preservation and preserve the antigenicity of connective tissues (Young et al., 1995; Nicolas et al., 1994; Hunziker, 1993; Keene and McDonald, 1993; Hunziker and Herrmann, 1987).
In the present study, immunological labeling was performed on tissues prepared by two procedures: (a) fresh material was treated either by ultra-rapid freezing or high-pressure freezing freeze-substitution, and resin embedding; and (b) chemically fixed material was submitted to freeze-fixation, freeze-substitution, and embedding in the same way as the freshly isolated material. We tested the latter procedure because, due to the way the worms were collected during oceanic missions, the majority of the worm tissues were accessible only after glutaraldehyde fixation. This procedure was also applied on fish scales to compare the influence of the glutaraldehyde prefixation on the immunolabeling of two types of fibrillar collagens: (a) the unbanded fibrillar collagen of the worm cuticle, an extremely long molecule with a globular domain (Gaill et al., 1994,1995; Mann et al., 1992; Murray and Tanzer, 1985), which has no counterpart in vertebrates (Har-El and Tanzer, 1993), and (b) the ubiquitous vertebrate Type I collagen, which is the main component of fish scales (Zylberberg et al., 1992a; Kimura et al., 1991).
Materials and Methods
Animals
Fixations
Postembedding immunolabeling after cryofixation and cryosubstitution of prefixed material was tested on specimens treated by glutaraldehyde. Pieces of the body wall from the vent worm R. pachyptila were fixed immediately after recovery on board ship by injecting a 3% glutaraldehyde, 0.1 M sodium cacodylate-buffered solution (pH 7.4; 1 hr at 4°C). They were stored in sodium cacodylate buffer until further treatment in the laboratory. Both solutions were brought to sea osmolarity by addition of NaCl as previously described (Gaill et al., 1991).
Scales of the goldfish were fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate [pH 7.4; 1 hr at room temperature (RT)] and stored in sodium cacodylate buffer before performance of cryotechniques.
The samples were immersed for 1 hr in a solution of 20% glycerol in buffer and then cryofixed by the slam-freezing method as described below; for high-pressure freezing no glycerol was added in the buffer.
Freeze-substitution
The frozen samples, stored in liquid nitrogen, were then transferred into the freeze-substitution medium at −90°C for 3 days as described previously (Nicolas, 1991), either in pure acetone or in acetone containing 2.5% osmium tetroxide. A molecular sieve (type 4A 1/16; Union Carbide, Rungis, France) was added to the medium, which was gradually rewarmed from −90°C to −30°C over a period of 3 hr and left at −30°C for 2 hr. Some samples were kept in acetone at −20°C and embedded in LR White at −20°C. After a final rewarming to reach RT with a step of 30 min at this temperature, the other samples were washed in the pure solvent and embedded either in LR White polymerized at 50°C or in Epon.
Postembedding Immunogold Labeling
All steps of the labeling procedure, except the primary antibody incubation overnight, were performed at RT.
Ultrathin sections were collected on gold grids. They were washed for 15 min in a blocking solution of PBS containing 0.5% BSA (bovine serum albumin) and 0.05% Tween 20. The sections were then incubated overnight at 4°C in 20-μl drops of the blocking PBS containing the primary antibodies at the appropriate dilution. After several washes in pure PBS and one more in the PBS-BSA-Tween 20 for 15 min, the sections were incubated in the secondary gold-conjugated antibody. The sections were then washed in PBS. The immunoreaction was stabilized by floating the grids on 0.2% glutaraldehyde in PBS for 2 min. After several rinses in PBS and in distilled water, they were contrasted in aqueous saturated uranyl acetate solution.
The Type I collagen was identified in the goldfish scales using as primary antibody a polyclonal antibody raised against the dermal pepsin-digested Type I collagen of the goldfish (provided by Dr. D. J. Hartmann, Institut Pasteur, Lyon). This antibody was the same as that previously tested (Zylberberg et al., 1992a). It was used at a concentration of 1:60 in the PBS-BSA-Tween 20 solution.
To control the specificity of the labeling, the primary antibodies were substituted with appropriate dilutions of nonimmune rabbit serum or PBS-BSA-Tween solution.
Results
Goldfish scales and worm cuticles were submitted to the same cryotechniques before postembedding immunolabeling.
Fish Scale Collagen
The basal plate of the goldfish is composed of thick collagen fibrils (about 100 μm in diameter) organized in superimposed layers parallel to the scale surface and arranged in a plywood-like structure. In each layer, the fibrils are parallel to each other and their direction varies from one layer to the other. In the goldfish basal plate, thin collagen fibrils called TC fibers (Onozato and Watabe, 1979) perpendicular to the scale surface cross the plywood-like structure. Thick and thin collagen fibrils of the basal plate are synthesized by the hyposquamal scleroblasts that form a continuous layer lining the basal surface of the scales.
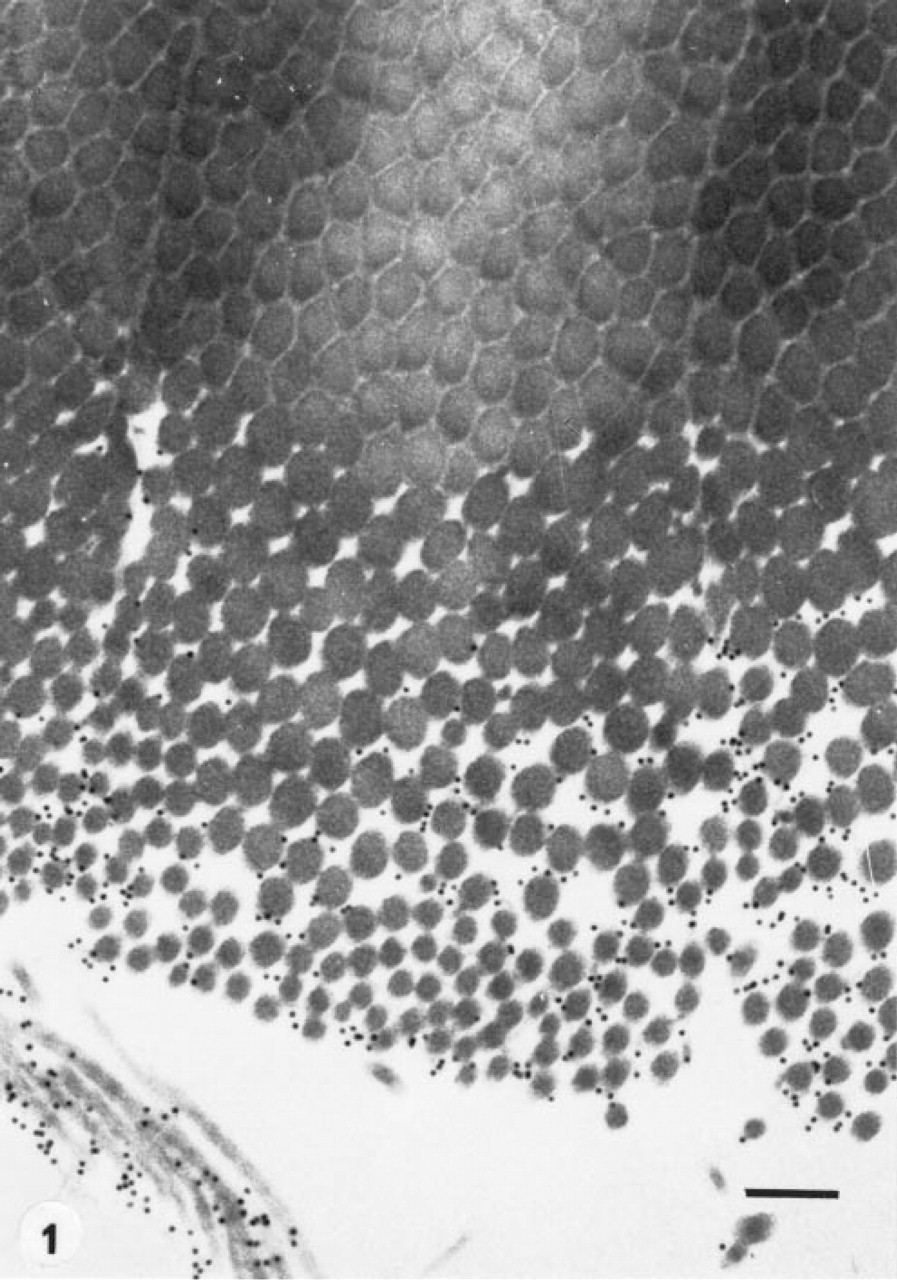
Goldfish scale. Pre-embedding immunolabeling after proteinase K disruptive treatment. Lower part of the basal plate. Note that only the collagen fibrils of the disrupted part are labeled by the gold particles, which do not penetrate within the compact tissue. Original magnification x 50,000. Bar = 200 nm.

Goldfish scale. Postembedding immunolabeling after freeze-substitution in OsO4-acetone, LR White. Type I collagen antibody-protein A-gold (10 nm).
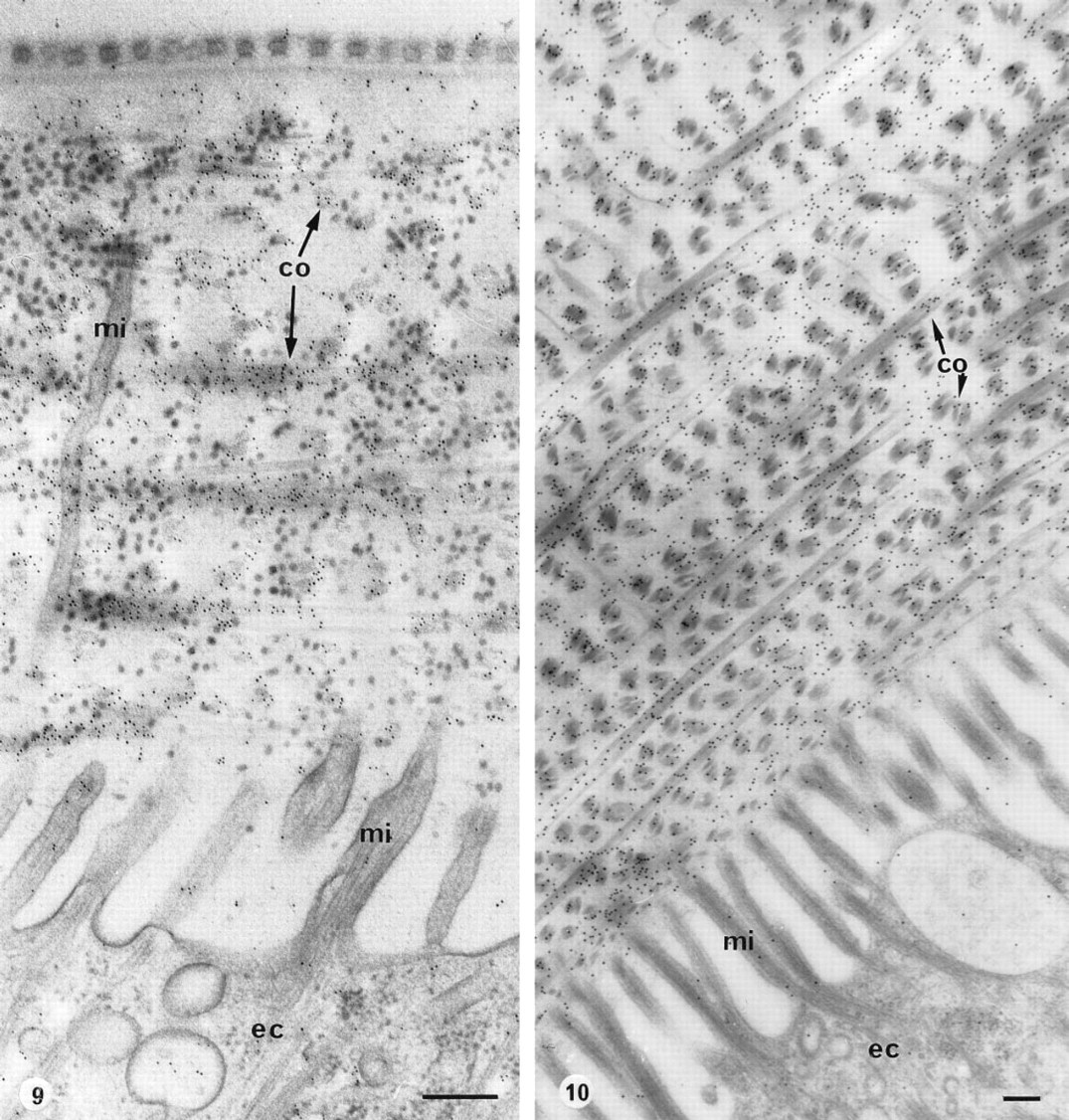
Harmothoe lunulata. Cuticle underlined by epidermal cells (ec) in the elytrum. Slam-freezing, freeze-substitution in OsO4-acetone. co, collagen; mi, microvilli.
Immunogold labeling using pre-embedding procedures was obtained only after disruptive treatments. These treatments disorganized the collagenous network, at least at the surface of the specimens. Only isolated collagen fibrils were decorated with gold particles when antibody against Type I collagen was used (Figure 1). No labeling was observed on the fibrils within the compact collagenous network. The scales that were cryofixed by slam-freezing, cryosubstituted with acetone supplemented with osmium tetroxide, and embedded in Epon showed improved preservation of the cells and of the collagen fibrils, as described elsewhere (Zylberberg and Nicolas, 1982). However, the immunolabeling of these specimens was not specific, even when osmium tetroxide was omitted during cryosubstitution. Specific labeling was obtained only in cryofixed samples cryosubstituted in acetone either with or without osmium tetroxide and embedded in LR White at both − 20°C and 50°C (Figures 2, 3, and 5). The gold particles were distributed over the basal plate on the thick collagen fibrils forming the plywood-like structure and on the thin TC fibers (Figure 2). No labeling was observed in the hyposquamal scleroblasts, which exhibit flat RER saccules (Figure 4). The labeling was not improved in specimens embedded in LR White at low temperature (Figure 5).
Harmothoe lunulata. Cuticle covering the elytrum. High-pressure freezing, freeze-substitution in OsO4-acetone, embedding in Epon, cuticular collagen antibody-protein A-gold (10 nm). The cuticular structure is very well preserved and specifically labeled. Original magnification x 30,000. Bar = 200 nm.
Scales submitted to high-pressure freezing and then to freeze-substitution in acetone containing osmium tetroxide and embedded in LR White showed a wellpreserved collagenous network, the labeling of which did not appear as intense as that obtained after slam-freezing (Figures 6 and 7).
No specific labeling was obtained with sections of goldfish scales that were chemically fixed before cryo-fixation by slam-freezing, cryosubstitution in acetone with or without osmium tetroxide, and embedding in Epon or in LR White.
No labeling was obtained on control sections submitted to nonimmune rabbit serum (Figure 8) or when the primary antibody was omitted.
Cuticular Collagen
The worm cuticle secreted by the epidermal cells is composed of unbanded fibrils organized in superim-posed layers, forming a plywood-like structure that is not as dense as that of the fish scale. The cuticle is crossed by microvilli that arise from the epidermal cells and reach the body surface.
For studying the coastal annelids, all laboratory facilities were available. Therefore, slam-freezing and cryosubstitution, which were known to preserve the ultrastructure and the antigenicity of the tissues (Nicolas, 1991), were applied on both species, H. lunulata and A. marina. Elytra of H. lunulata were freeze-substituted with acetone-osmium tetroxide and embedded in either LR White or Epon. High specific labeling of the collagenous network was obtained with both resins (Figures 9 and 10). Low-temperature embedding did not improve the intensity of the labeling. Moreover, the labeling of the cuticular collagen was obtained only after a cryosubstitution with osmium tetroxide added to the medium; no labeling was observed after the use of pure acetone. The same results were obtained with A. marina embedded in Epon.
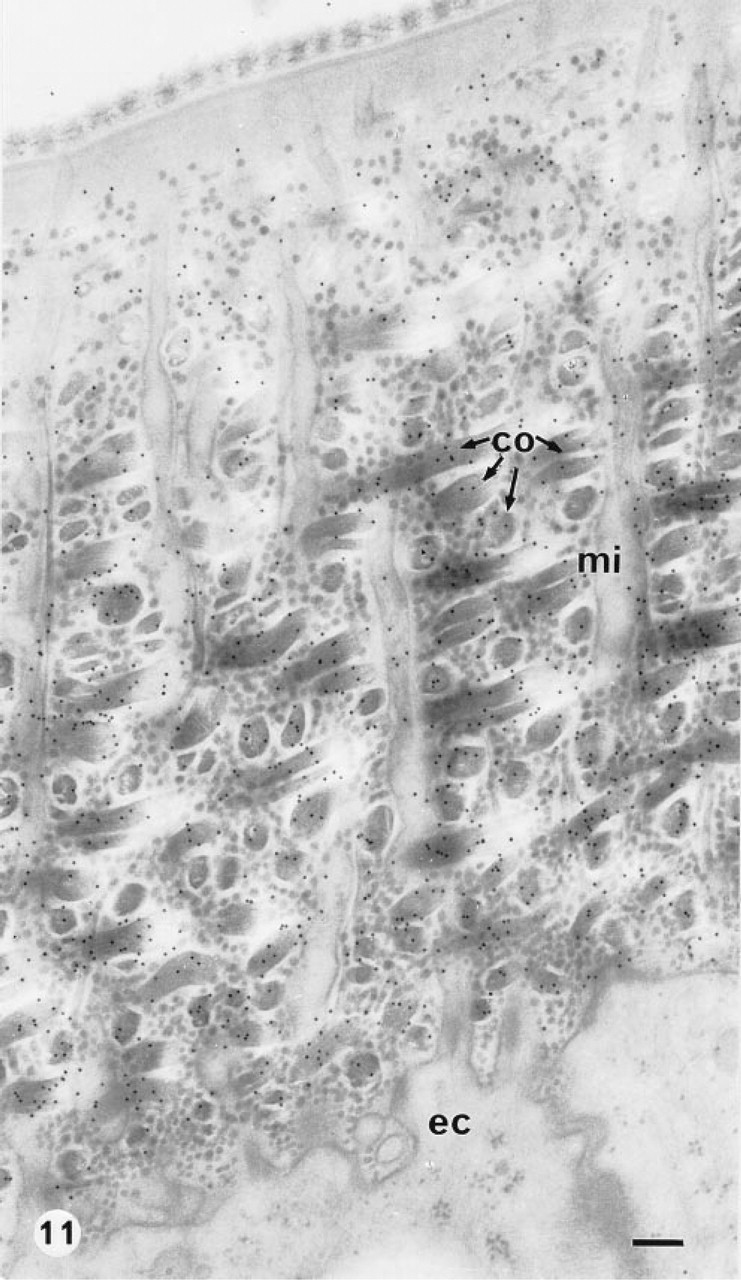
Immunolabeling of the cuticular collagen after high-pressure freezing followed by cryosubstitution in acetone-osmium tetroxide and embedding in Epon was tested on the elytra of H. lunulata (Figure 11). With the use of high-pressure freezing, extractions were observed in the ground cytoplasm and the vesicles were swollen, whereas the preservation of the cuticle ultrastructure was improved. However, the immunolabeling did not appear as intense as that obtained with slam-freezing (Figure 9).
The specimens of R. pachyptila were cryofixed on shipboard by plunging in liquid propane. Few results were obtained because of the difficulty of reproducing good quality freeze-fixations without specialized devices, and only small, shallow areas appeared to be well preserved. In these areas the collagen fibrils organized in an orthogonal plywood-like structure were well preserved and were labeled with gold particles (Figure 12).
Riftia pachyptila, body cuticle.
Results obtained after the different procedures used a
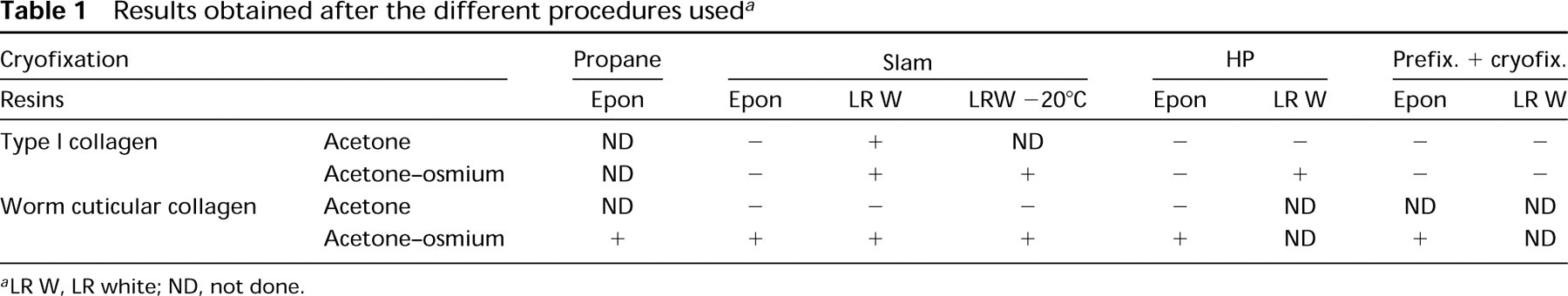
aLR W, LR white; ND, not done.
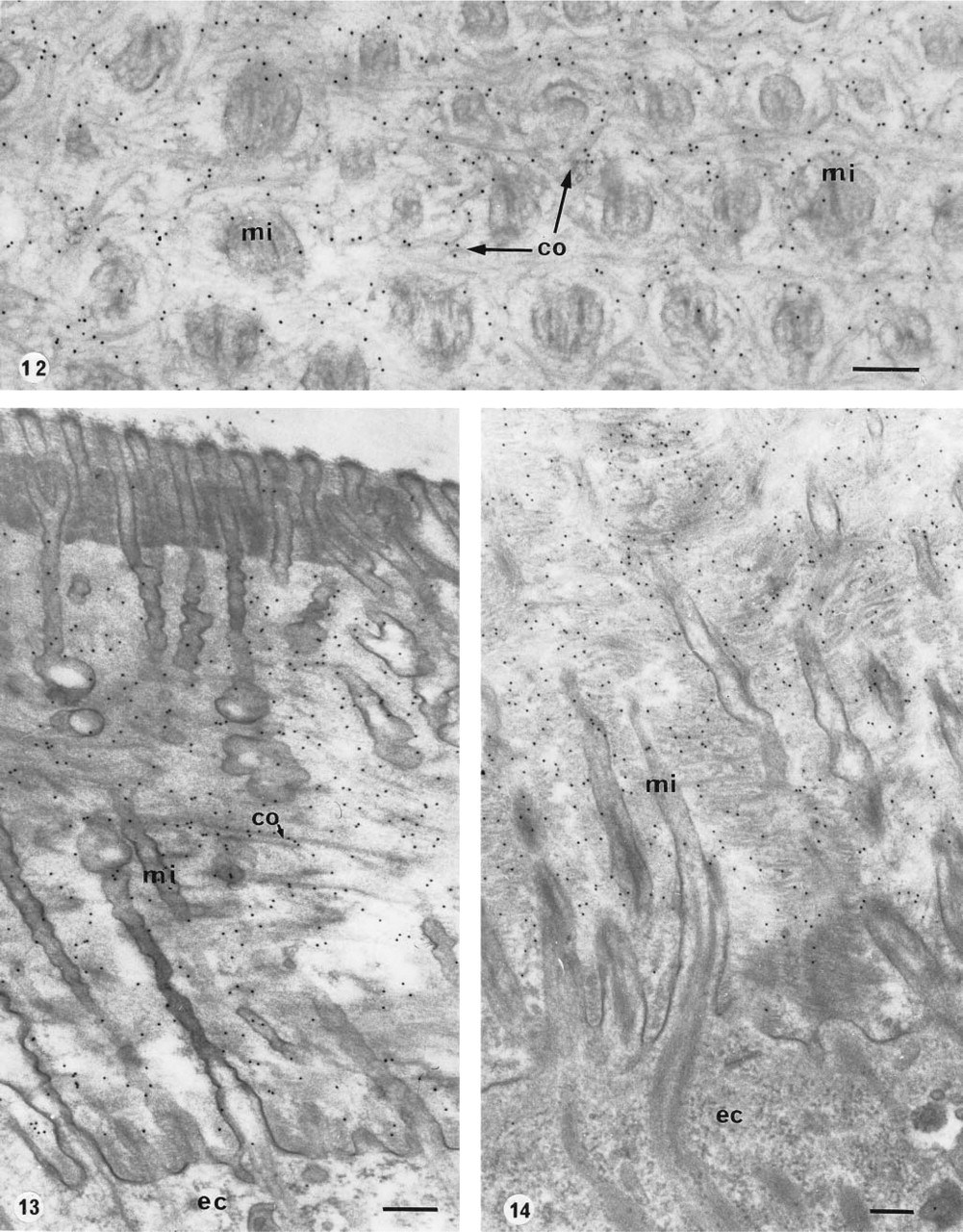
Most of the tissues collected during the cruise were accessible after fixation by aldehydes only, mostly glutaraldehyde, which is known to inhibit immunolabeling reactions. Nevertheless, we carried out immunolabeling techniques after freeze-fixation of these chemically fixed tissues. Slam-freezing (Figure 13) and high-pressure freezing (Figure 14) were used. The samples were freeze-substituted in a medium containing osmium tetroxide and embedded in Epon. The freeze-fixation of chemically pre-fixed and cryoprotected specimens ensures the preservation of a thicker area (about 200 μm with slam-freezing) from the freezing front than that obtained with fresh material without cryoprotection (about 10–15 μm). The high-pressure freezing produced some swelling of the cytoplasmic vesicles, similar to that observed in non-pre-fixed material. The collagenous network was well preserved and abundant gold particles were specifically distributed on the collagen fibrils (Figure 14).
Control sections submitted to the nonimmune rabbit serum or to PBS-BSA-Tween solution were not labeled.
The results are summarized in Table 1. After the slam-freezing method, we tested two cryosubstitution media, pure acetone or acetone-osmium, and the two resins, Epon or LR White. This latter was polymerized at −20°C or 50°C. The use of LR White polymerized at low temperature did not improve the immunolabeling compared to that obtained at 50°C. Therefore, for the other experiments the polymerization was achieved at 50°C. It appeared that the best compromise for characterization of Type I collagen was obtained after cryosubstitution with acetone-osmium and embedding in LR White, whereas for cuticular collagen it was obtained after cryosubstitution in acetone-osmium and embedding either in Epon or LR White.
Discussion
The present study has shown that immunoelectron microscopy of fibrillar collagens in compact tissues is achieved using postembedding labeling after freeze-fixation and freeze-substitution, which also yields good preservation of the spatial architecture of the collagenous network in the worm cuticle and in the fish scale. Previous studies using the same antibodies have shown that postembedding immunolabeling has failed after chemical fixation in worm cuticle (Hamraoui, 1994) and in fish scales (unpublished results). These results are consistent with the fact generally admitted that conventional techniques—chemical fixations, dehydration, and embedding—produce alterations that lead to masking of the antigen.
In vertebrates, collagen epitopes are highly sensitive to aldehydes contained in the fixatives and particularly to glutaraldehyde, the most successful aldehyde used for electron microscopy (Young et al., 1995; Hunziker, 1993), these aldehydes showing a high affinity for α-helices (Hayat, 1981). The formation of intermolecular crosslinks is shown by changes in the staining patterns of the fixed fibrillar collagens (Bairati et al., 1968). Cryotechniques offer advantages because they minimize modifications of the morphological features and preserve antigenicity (Hunziker, 1993). Slam-freezing of fresh worm cuticle and fish scale has succeeded in providing well-preserved structures within the cells as well as in the compact extracellular matrix. High-pressure freezing of fresh tissues has improved the structural preservation of the compact extracellular matrix but not its immunoreactivity, as observed in another extracellular matrix (Nanci et al., 1994). It has induced some artifacts in the cell structures, such as swelling of the vesicles and extraction of the ground cytoplasm. Such artifacts were believed to occur during specimen mounting before exposure to high-pressure freezing (Kiss and Staehelin, 1995).
Fine preservation of the structures as well as intense immunolabeling was obtained when osmium tetroxide was added to acetone, the freeze-substitution medium, although osmium tetroxide is known to destroy antigenicity by protein-protein crosslinking (Newman and Jasani, 1984; Nielson and Griffith, 1979). Moreover, osmium tetroxide must be added to the freeze-substitution medium to preserve the antigenicity of the worm cuticular collagen. In our study, we have found that the antigenicity of the compact collagenous network of worm cuticle was also preserved in chemically fixed specimens subsequently freeze-fixed either by slam-freezing or by high-pressure freezing and freeze-substitution. These positive results suggest that the main cause of labeling loss of the worm cuticular collagen is not due to the aldehyde fixation but to the other procedural steps. The dehydration at room temperature might be a disturbing step, as it induces osmotic changes and shrinkage (Oprins et al., 1994; Hayat, 1970). Freeze-substitution, which consists of the withdrawal of water in a solid state, results in a very slow and gentle dehydration, which reduces the possibility of ultrastructural changes to a minimum (Chan et al., 1993; Yamashita and Yasuda, 1992).
In contrast, the immunoreactivity of the Type I collagen of the fish scale was not preserved in samples fixed before freeze-fixation and freeze-substitution. On the other hand, unlike the worm cuticular collagen, which is immunolabeled after embedding in LR White and in Epon, the Type I collagen of fish scale is immunoreactive only in specimens embedded in LR White, a hydrophilic acrylic resin that provides good access to epitopes (Brorson et al., 1994; Griffiths, 1993; Newman and Hobot, 1993). No labeling was obtained in samples embedded in Epon. This might be explained by the fact that this resin could co-polymerize with the tissue, forming covalent bonds (Kellenberger et al., 1987) and hampering immunolabeling.
The failure of labeling of the collagenous network in the fish scales while the worm cuticle remains immunoreactive can be related to the presence of noncollagenous components such as those involved in the mineralization of the fish scales (Zylberberg et al., 1992b; Zylberberg and Nicolas, 1982), which prevent access to Type I collagen epitopes. Immunolabeling might also be hampered by the higher compactness of the plywood-like structure in the fish scales than in the cuticle. Alternatively, it is possible that epitopes of the cuticular collagen are less sensitive to aldehydes than those of Type I collagen of vertebrates. Epitopes of the cuticular collagen are not destroyed by chemical fixation and become accessible to antibodies after freeze-fixation and freeze-substitution.
Fixation is considered to be the most important of the many steps involved in the processing of specimens for immunoelectron microscopy (reviewed by Williams and Faulkner, 1993). Nevertheless, freeze-substitution and embedding appeared to be important also, since the substitution medium and the resin could also prevent the labeling of antigen as observed in the present study.
Our results are consistent with
Footnotes
Acknowledgements
This work was supported by grants from the Centre National de la Recherche Scientifique (CNRS) URA 1488, UPR 9042, URA 1137.
We are indebted to Dr. Aries Kovoor (Université Paris 7 and CNRS) for the English correction of the manuscript. We also acknowledge the excellent service of the photographic department of the CIME.