
Abstract
Genetic and molecular studies in humans and mice indicate that Runx2 (Cbfa1) is a critical transcriptional regulator of bone and tooth formation. Heterozygous mutations in Runx2 cause cleidocranial dysplasia (CCD), an inherited disorder in humans and mice characterized by skeletal defects, supernumerary teeth, and delayed eruption. Mice lacking the Runx2 gene die at birth and lack bone and tooth development. Our extended phenotypic studies of Runx2 mutants showed that developing teeth fail to advance beyond the bud stage and that mandibular molar organs were more severely affected than maxillary molar organs. Runx2 (−/−) tooth organs, when transplanted beneath the kidney capsules of nude mice, failed to progress in development. Tooth epithelial-mesenchymal recombinations using Runx2 (+/+) and (−/−) tissues indicate that the defect in mesenchyme cannot be rescued by normal dental epithelium. Finally, our molecular analyses showed differential effects of the absence of Runx2 on tooth extracellular matrix (ECM) gene expression. These data support the hypothesis that Runx2 is one of the key mesenchymal factors that influences tooth morphogenesis and the subsequent differentiation of ameloblasts and odontoblasts.
Keywords
I
Humans with mutations in RUNX2 (CBFA1) exhibit cleidocranial dysplasia (CCD), an autosomal dominant disorder defined by several skeletal and dental defects. Driven by an interest in understanding the pathogenesis involving dentition in human CCD, we recently described the patterns of Runx2 expression during normal mouse tooth development. Our data indicated that Runx2 expression is spatially restricted to dental papilla mesenchyme at that stages precede crown development and is markedly downregulated after morphogenesis is complete (D'Souza et al. 1999). Interestingly, Runx2 expression persists in dental follicle mesenchyme and the periodontal ligament during the pre-eruptive phase of tooth development (Bronckers et al. 2001). Our phenotypic analysis of tooth development in Runx2 (−/−) mice was limited to the neonatal stage and revealed an arrest at the early cap stage (D'Souza et al. 1999). These results raise intriguing questions about the role of this transcription factor in tooth morphogenesis. Questions remain about the exact stage of tooth arrest in Runx2 (−/−) mice and whether maxillary and mandibular molars and incisors are affected with the same degree of severity. Furthermore, it is unclear whether the phenotype of tooth arrest is a result of a marked delay or a true cessation of tooth morphogenesis and whether the arrest is caused by a defect in dental mesenchyme that subsequently affects epithelial patterning. Finally, the functional consequences of the lack of Runx2 on terminal differentiation of odontoblasts, cells that closely resemble osteoblasts, are not known.
The objectives of these studies were to provide further insights into the alterations in dentition of Runx2 (−/−) mice. We have extended our phenotypic studies of tooth development in Runx2 mutant mice using histomorphometric approaches. In addition, we have evaluated the developmental capacity of Runx2 mutant tooth organs when transplanted beneath the kidney capsules of nude mice and have performed cultures of tooth epithelial-mesenchymal recombinations using Runx2 (+/+) and (−/−) tissues. Finally, to assess the phenotypic changes in cytodifferentiation, we performed molecular assays for the study of tooth extracellular matrix (ECM) gene expression. Collectively, our data support the conclusion that Runx2 is a key mesenchymal factor that influences the morphogenetic patterning of dental epithelium and the subsequent morphodifferentiation of ameloblasts and odontoblasts, cells responsible for the formation of enamel and dentin matrices, respectively. Intriguingly, the absence of Runx2 showed differential effects on the expression levels of dentin matrix genes.
Materials and Methods
Tissues for Histological and Molecular Analyses
Breeding pairs of mice heterozygous for Runx2 were initially obtained from Dr. Michael Owen (London, UK) and was used to generate Runx2 wild-type (+/+), heterozygous (+/-) and homozygous mutant (−/−) embryos for the studies described below. Original reports describing the phenotype of Runx2 homozygote-null mutants used the identical strain of mice (Otto et al. 1997). PCR genotyping was performed on tail DNA using a cocktail of three primer sets designed in the laboratories of Dr. Bjorn Olsen (Boston, MA) under conditions described earlier (D'Souza et al. 1999). To extend the mouse line, NMRI females were mated with C57/BL6 Runx2 (+/-) males. Using vaginal plug times as E0 (embryonic day 0), embryos staged from E12 to day 0 (newborn) were collected. DNA genotyping for this set of breedings was performed with newly designed sets of primers directed against the wild-type and mutant Runx2 alleles as follows: 5′AAG ATG GAT TGC ACG CAG GTT CTC 3′ and 5′CAC GGA GCA CAG GAA GTT GGG A 3′; 5′TGA GCG ACG TGA GCC TGG 3′ and 5′CAC GGA GCA CAG GAA GTT GGG A 3′. For every embryo/pup genotype, results were correlated with phenotypic changes noted by gross examination. Tissues were fixed overnight in 4% paraformaldehyde (PFA) and processed routinely for embedding in paraffin. Serial sections 7 μm thick were stained with H&E for histologic analysis. For in situ hybridization, sections representing the entire thickness of the alveolar segment were selected.
Tissue Culture and Kidney Capsule Transplants
Tissue recombinations between dental epithelium and mesenchyme were done as previously described (Vainio et al. 1993). E14 Runx2 (−/−) mandibular molar epithelium and mesenchyme were separated and recombined with E13 wild-type tooth tissues. Recombinant tissues were cultured for 5 days under normal culture conditions and processed for histology. Using methods described by Kratochwil et al. (1996), mandibular first molar segments were carefully dissected at E13 and E14 from Runx2 (+/+) and (−/−) embryos and transplanted beneath the renal capsule of young adult nude mice. After 3 weeks, animals were sacrificed and kidneys dissected for histological processing and microscopic analysis. A total of 16 mutant and 11 wild-type transplants were cultured.
In Situ Hybridization
Maxillae and mandibles were dissected from E18.5 and Day 0 Runx2 (+/+) and Runx2 (−/−) mouse embryos and pups and were immediately fixed by immersion in buffered 5% paraformaldehyde. Tissues were processed routinely for paraffin embedding without prior demineralization, and sagittal sections, 5-7 μm thick, were mounted on silane-coated glass slides. To study extracellular matrix gene expression, [α-35S]-UTP-labeled sense and antisense riboprobes were generated to murine proα1(I) collagen (Metsaranta et al. 1991), dentin sialophosphoprotein Dspp and dentin matrix protein 1-Dmp1 (D'Souza et al. 1997), osteocalcin-Ocn (Ducy et al. 1997), and ameloblastin-Am (Lee et al. 1996). In situ hybridizations were performed under conditions of high stringency as previously described (D'Souza et al. 1997). After post-hybridization treatments, sections were counter-stained with hematoxylin and images digitized using an Olympus DP-10 photographic system.
RT-PCR Analysis of Dentin ECM Gene Expression
The effects of the absence of Runx2 on the expression levels of two of the most dominant dentin ECM genes, type I collagen and Dspp, were studied further at E14 and day 0 using RT-PCR. Mandibular incisor and first molar organs were carefully microdissected from embryonic stage 14 (E14) and day 0 (newborn) Runx2 (+/+) and (−/−) mice. Molar organs were also obtained from postnatal day 21 mandibles of Runx2 (+/+) mice. Tissues were immediately snap-frozen in liquid nitrogen and total RNA extracted using RNA STAT-60 (Tel-Test; Friendswood, TX). After cDNA synthesis using oligo (dT) primers and reverse transcriptase, RT-PCR analysis was performed using the following primer sets: α1(I) collagen, 5′TCCTGCTCCTCTTAGGGG 3′; 5′CAA-CAGCACCATCGTTGC 3′ and glyceraldehyde 3 phosphate dehydrogenase (GAPDH) −5′CGTCCCGTAGACAAAATGGT 3′; 5′TTCCCGTTCAGCTCTGGGAT 3′, respectively. Dspp-specific primers and the PCR conditions for the amplifications of these genes have been previously described (D'Souza et al. 1997). Amplification products were analyzed on a 1.5% Agarose gel and visualized by ethidium bromide staining.
Results
Our previous analysis of the Runx2 mutant phenotype was preliminary and was performed on restricted material (D'Souza et al. 1999). We now performed a more detailed analysis using serial sections and several stages.
Runx2 Is Needed for the Progress of the Molar Organ from the Bud to the Cap Stage
Frontal sections of the molar region of Runx2 (−/−) embryos and their wild-type littermates were examined at developmental stages of E12, E13, E14, E16, and E18. In Runx2 (+/+) dentition, no major differences were detected between the development of upper and lower molars. At E12, the wild-type and Runx2 mutant tooth germs were morphologically similar (not shown). The first difference was observed during the early bud stage at E13, when molar tooth buds of the Runx2 (−/−) mutants were slightly delayed in development. Compared to Runx2 (+/+) molar organs that had reached the full cap stage, maxillary molar development appeared delayed in Runx2 mutant embryos.
Runx2 Mutant Molar Organs Show the Presence of Accessory Buds
By stage E14, the developmental arrest was remarkable in both upper and lower molars (Figure 1B). Although the dental mesenchymal condensate appeared normal in Runx2 (−/−) molar organs, the enamel knot was not visible as a morphologically distinct entity within the ectodermal compartment. A characteristic feature of the upper molar epithelium at E14 was the presence of an extra budding at the palatal (lingual) side of the bud (Figure 1B and 1C). The morphogenesis of Runx2 mutant molars did not proceed during subsequent embryonic development. However, at E16 extra buddings were more apparent in the anterior and palatal aspects of upper molars. These aberrant outgrowths were sometimes observed in lower molars (Figures 1E and 1F). At E18, the tooth buds of mutant molars had regressed and the actual tooth buds could not be easily identified within the many epithelial ingrowths (Figures 1H and 1I).
Results of our more detailed histologic analyses revealed that Runx2 is needed for the bud-to-cap stage transition and for the development of a morphologically distinct enamel knot. The absence of functional Runx2 causes failure to form the epithelial cervical loops and mesenchymal dental papilla. In addition, Runx2 apparently prevents epithelial budding at the lingual aspect of wild-type tooth germs.
Absence of Runx2 Affects Development of Mandibular Molars More Severely Than Maxillary Molars
As Figure 1H indicates, the dental lamina in E18 Runx2 (−/−) mandibles appear highly proliferative, with no clear advancement to the cap stage. This dysmorphology sharply contrasts with the appearance of the Runx2 mutant maxillary molar organ that progresses to the bud stage.
Other Craniofacial Abnormalities in Runx2 (−/−) Mice
In a majority of Runx2 (−/−) embryos studied a failure of fusion between the shelves of the secondary palate was noted (Figures 1J and 1K). The palatal clefting appeared as an isolated defect and was not associated with defects in primary palate fusion or cleft lip. In wild-type mice embryos, eyelids are open at E15 and close from E16 until approximately 2 weeks after birth. Interestingly, in several Runx2 (−/−) embryos studied between E15 and the neonatal stage, eyelids showed a failure of closure (Figure 1L). These pheno-typic changes in the palate and eyelids helped to distinguish Runx2 (−/−) embryos/pups from their (+/+) and (+/-) littermates.
The Primary Defect in Runx2 (−/−) Tooth Organs Resides in the Dental Mesenchyme and Cannot be Rescued In Vivo
To assess if the defect is in the Runx2 (−/−) epithelium or the mesenchymal tissue, recombinations were performed. Dental epithelium and mesenchyme from both Runx2 (+/+) and (−/−) mandibular E13 molars were recombined in different combinations and explants cultured for 6-8 days, as described earlier. When epithelial and mesenchymal tissues from Runx2 (+/+) tooth organs were separated and recombined for culture, they developed into a multicusped bell-stage morphology (data not shown). When Runx2 (−/−) mesenchyme was combined with (+/+) epithelium, no obvious development occurred; only some ingrowths of the epithelium were seen (6/8 explants; Figure 2A). When Runx2 (+/+) mesenchyme was cultured with mutant epithelium, morphogenesis appeared normal and bell-stage molar morphology was evident (2/6 explants) (Figures 2B and 2C). Hence, the primary defect in the Runx2 (−/−) tooth organ is within the dental mesenchyme. Because Runx2 (−/−) mice succumb at birth, we transplanted mandibular first molar organs from E13 and E14 mutant embryos beneath the kidney capsule of nude mice to examine if their development would be rescued under long-term culture in vivo. However, only cyst-like structures formed, which were filled with keratin, and no teeth were observed (Figure 2D). In addition, two wild-type and one mutant tooth germ were cultured under kidney capsule for 2 weeks. Two well-mineralized late bell-stage teeth were formed with surrounding alveolar bone, but the mutant tooth had regressed (Figure 2E). A total of 14 Runx2 mutant and 10 wild-type transplants were performed.
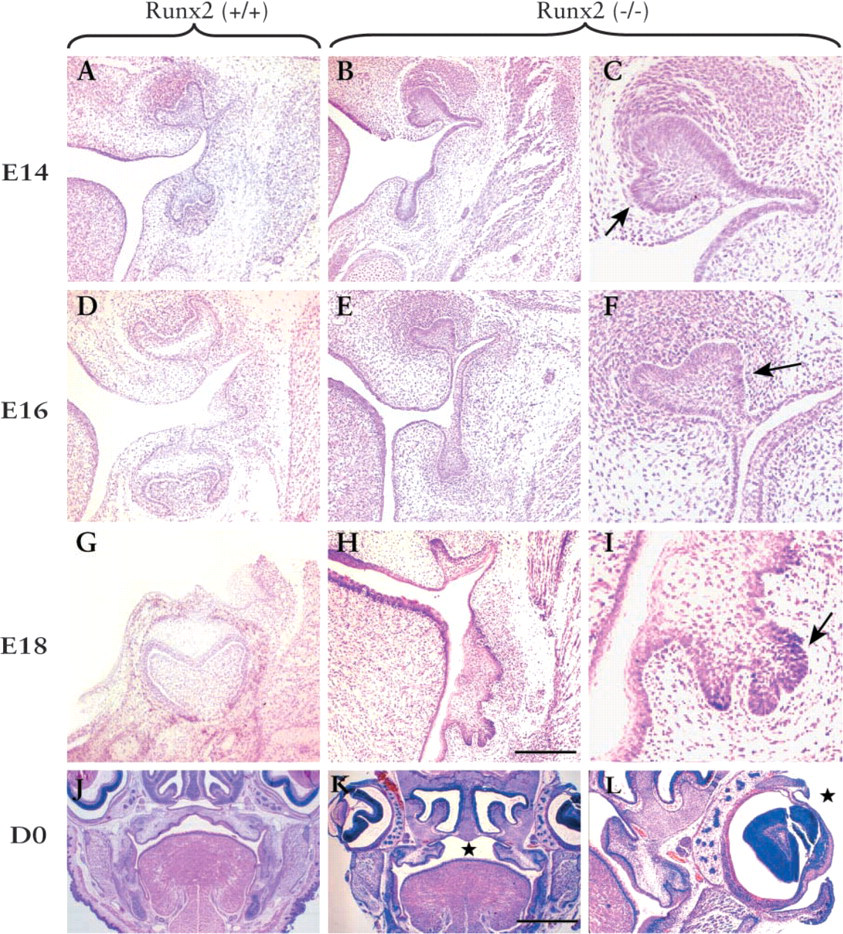
Histological analysis of Runx2 (−/−) phenotype with wild-type as control. All sections are cut frontally and stained with H&E. (
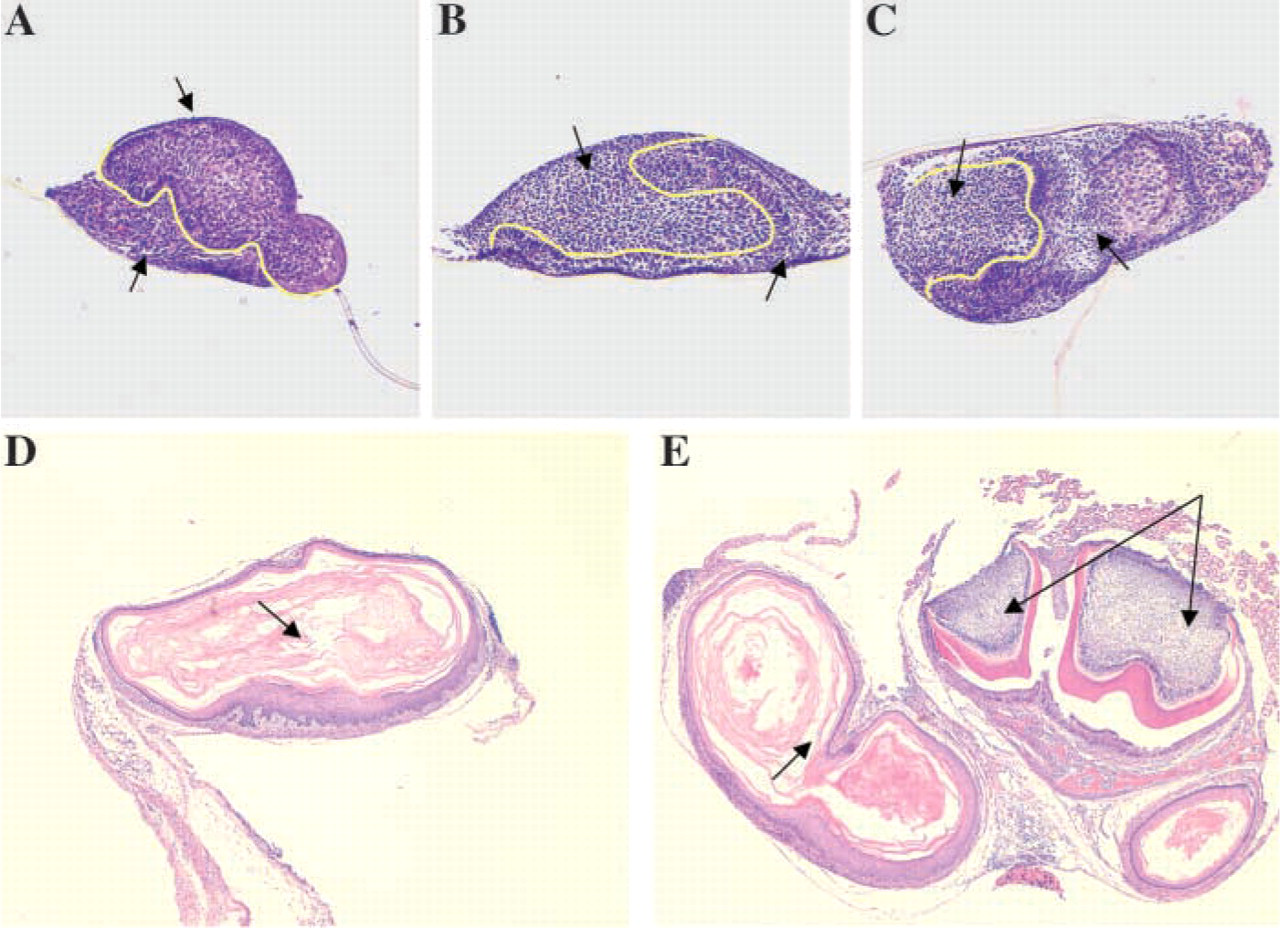
Tissue recombinations with Runx2 (+/+) and (−/−) molar organs (
Differential Effects on Tooth ECM Gene Expression
Because incisors were less severely affected than molar organs in Runx2 (−/−) mice (D'Souza et al. 1999), we were able to study ECM gene expression in putative odontoblasts and ameloblasts. In all sections of Runx2 (−/−) maxillary and mandibular incisor organs studied, putative odontoblasts and ameloblasts appeared poorly differentiated from a morphological standpoint. Cells were cuboidal rather than columnar and lacked nuclear polarity. In situ hybridization results showed hybridization for α1(I) collagen gene expression in newly differentiated odontoblasts as well as in osteoblasts in surrounding bone of newborn Runx2 (+/+) incisors (Figure 3A). Runx2 (−/−) incisors showed α1(I) collagen labeling in putative odontoblasts but not in surrounding connective tissue, where osteogenesis would normally occur (Figure 3B). As expected, a strong signal for Dspp was seen in newly differentiated odontoblasts and a group of ameloblasts within an Runx2 (+/+) incisor (Figure 3C). In sharp contrast, lower levels of Dspp expression appeared localized to a cluster of putative odontoblasts in an Runx2 (−/−) incisor (Figure 3D). Expression of Dmp1 and OCN appeared markedly downregulated in mutant dental mesenchyme compared to normal littermates. Because the underlying defect in Runx2 (−/−) dental mesenchyme may alter the fate of ectodermal cell differentiation into ameloblasts, we evaluated expression of an ameloblast-specific gene marker, ameloblastin (AM). AM transcripts were visible in putative Runx2 (−/−) ameloblasts and the level of expression was comparable to that noted in a wild-type incisor.
RT-PCR analysis was performed to examine whether the absence of Runx2 affected the expression levels of two major dentin ECM genes, ct1(I) collagen and Dspp. Figure 4A shows no differences in levels of ct1(I) collagen transcripts in E14 Runx2 (−/−) molar and incisor organs compared to wild-type littermate controls. At this stage, before the terminal differentiation of odontoblasts in Runx2 (+/+) molars or incisors, Dspp expression is negligible. In Runx2 (−/−) tooth organs, Dspp transcripts are barely visible. A positive control sample of a 21-day-old molar tooth organ showed high levels of Dspp expression. In day 0 molar and incisor Runx2 (−/−) organs, ct1(I) collagen gene expression appeared unaltered. In contrast, Dspp transcripts are faintly detectable in mutant molars and incisors compared to Runx2 (+/+) tooth organs, in which high levels of Dspp expression were evident. Expression of GAPDH, a housekeeping gene used as an internal control, remained constant in all RNA samples analyzed in these studies (Figures 4A and 4B).
Discussion
Analysis of serial sections of Runx2 (−/−) mutant tooth organs revealed failure to advance fully past the bud stage of development. The lack of functional Runx2 disrupted the formation of a morphologically distinct enamel knot and the development of the cervical loop extensions of the ectodermal compartment. Hence, tooth morphogenesis appeared arrested in the transition from the bud to the cap stage.
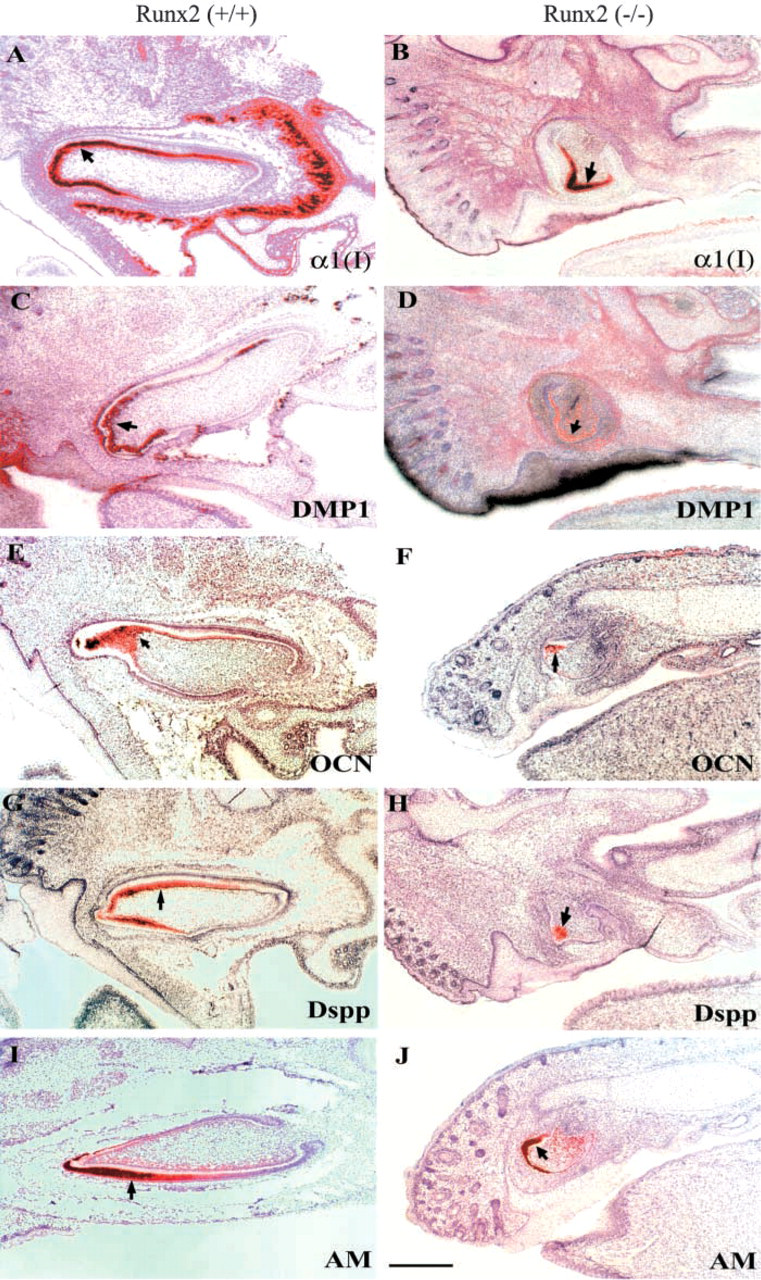
Brightfield digitized images of in situ hybridization comparing ECM gene expression in Runx2 (+/+) and (−/−) incisor organs. Arrows point to odontoblasts (
An interesting finding was the development of epithelial buddings, particularly in the maxillary molars. This suggests that the normal function of Runx2 may be to prevent their formation, by either controlling the rate of cell proliferation or the extent of apoptosis. They were seen on the palatal aspect of the tooth germ. This is intriguing, because, in humans and other mammals, succedaneous dentition arises as palatal/lingual extensions of the primary dental lamina. We propose that the extra buddings may have occurred at sites in which the formation of the secondary teeth is normally prevented in mice. This observation in the mouse genetic model of CCD can explain the presence of multiple supernumerary teeth seen fairly consistently in humans affected by the haploinsufficiency of Runx2.
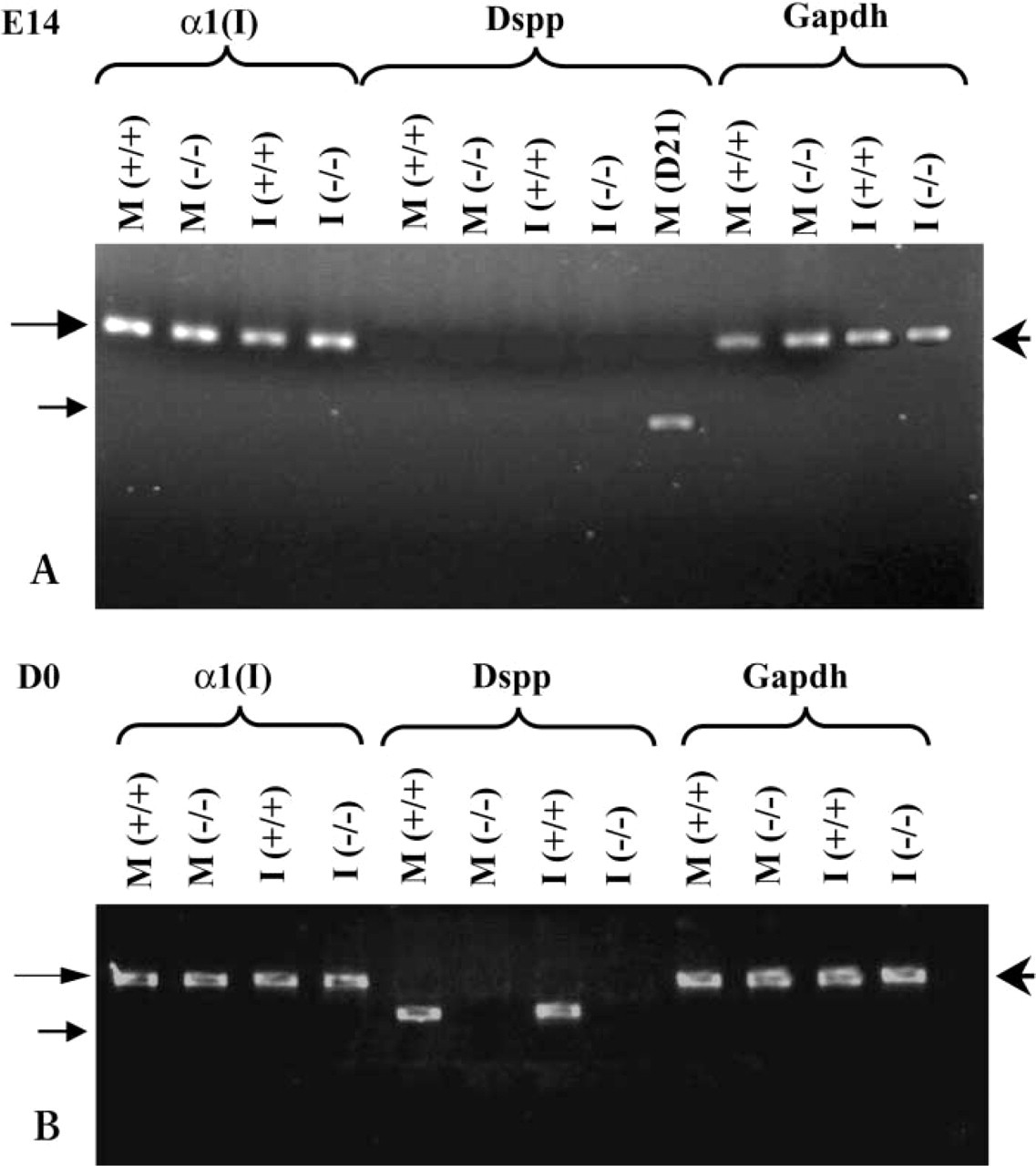
RT-PCR analysis of matrix gene expression in Runx2 (+/+) and Runx2 (−/−) molar (M) and incisor (I) organs. (
Our extended phenotypic analysis revealed that molars were more severely affected than incisors and that mandibular tooth organs were more phenotypically altered in the absence of Runx2 than their maxillary counterparts. Regional differences in the molecular regulation have been previously noted between the upper and lower molars. In Dlx1/Dlx2 double mutants, only maxillary molars fail to develop (Qiu et al. 1997; Thomas et al. 1997), and activinβA mutant mice have a phenotype opposite to it, with only mandibular molars and incisors affected (Ferguson et al. 1998). Our in situ hybridization analysis of Runx2 expression during normal tooth development did not show significant variation in spatial patterns of expression. It is possible that compensation for the lack of Runx2 by other Runx isoforms may affect the degree of severity of the tooth phenotype in different regions of the developing arches. However, of the other two known Runx2 isoforms, Runx1 expression is confined to dental epithelium and Runx3 is present at weak levels in mesenchyme (Yamashiro et al. 2002).
To the best of our knowledge, the findings of a cleft palate and early opening of the eyelids in Runx2 mutant embryos have not been previously described. Earlier phenotypic studies of Runx2 mutant embryos and newborn pups did not describe effects on palate or eyelid formation, suggesting that these phenotypic variations may be the result of incomplete penetrance (Komori et al. 1997; Otto et al. 1997). Interestingly, Yamachika et al. (2001) report a heterozygous C-to-T transition mutation within the runt domain of RUNX2 in a patient with cleidocranial dysplasia who also reported a cleft lip. Whether Runx2 plays a direct or indirect role in palate formation is an issue that remains unresolved. In the latter case, the failure of the palatal shelves to fuse may be secondary to the lack of bone formation that results in a hypoplastic mandible and the failure of the tongue to drop within the oral cavity. Previous studies have described the effects of epidermal growth factor (EGF) and the transforming growth factor-alphas (TGF-αs) in causing precocious opening of the eyelids in newborn mice (Smith et al. 1985; Berkowitz et al. 1996). Other studies have implicated tumor necrosis factor-α (TNF-α) converting enzyme (TACE/ADAM17) in regulating the availability of the EGF receptor ligand in vivo. Mice heterozygous for Tace and homozygous for an impaired EGFR allele were born with open eyes significantly more often than their wild-type littermates (Sunnarborg et al. 2002). Clearly, more studies are needed to study whether Runx2 is involved in the opening and closure of eyelids and its relationship with the EGF and TGF-α signaling pathway.
An additional goal of this study was to advance our understanding of the role of Runx2 in tooth cytodifferentiation events. Specifically, we sought to determine whether Runx2 influenced odontoblast terminal differentiation events, as measured by morphological criteria and the expression of key dentin matrix genes. Although Runx2 mutant molars arrested before the cap stage, incisors were less severely affected. In the latter, phenotypic changes seen at day 0 involved abnormal odontoblast morphodifferentiation and a highly disorganized and reduced layer of dentin matrix (D'Souza et al. 1999). Therefore, it is logical to conclude that the transcription factor plays a critical and non-redundant role in events that lead to cell differentiation at the late bell stage of incisor development.
Our strategy in assessing the molecular basis of the abnormal odontoblast and dentin phenotype was to focus on dentin ECM gene expression since Runx2 binding sites have been identified in the promoter regions of multiple genes that encode for matrix proteins that are common to bone and dentin (for review see Tsuji et al. 1998; Ducy 2000; Komori 2000; Kern et al. 2000). Although α1(I) collagen is a major gene product of odontoblasts, it is less specific for the odontoblastic phenotype because it is also expressed in other mineralizing cells, such as osteoblasts and cementoblasts. RT-PCR and in situ hybridization analyses revealed that, despite the arrest in tooth development, putative odontoblasts in Runx2 (−/−) incisor organs were able to transcriptionally activate the α1(I) collagen gene. Interestingly, type I collagen gene expression was shown to be absent in ossification zones of Runx2 (−/−) mice (Ducy et al. 1997; Komori et al. 1997; Hoshi et al. 1999). More recently, electrophoretic mobility shift assays and mutagenesis studies have definitively shown that Runx2 is a transcriptional regulator of osteoblast-specific expression of α1(I) and α2(I) collagen genes. Cell-specific differences in type I collagen gene regulation have been previously documented in the Mov13 mouse strain that was generated with a retroviral insert in the α1(I) collagen gene. Osteoblasts transcribed the mutant allele at significantly lower levels, while odontoblasts transcribed the mutant α1(I) collagen allele at normal levels. In other type I collagen-expressing mesodermal cells, expression of the mutant α1(I) collagen allele was completely blocked (Kratochwil et al. 1989,1993).
The downregulation of Dspp, Dmp1, and OCN seen in Runx2 mutant incisor organs can be interpreted as an arrest in the maturation of odontoblasts. Taken together, our phenotypic analysis suggests that the absence of Runx2 affects the terminal phases of odontoblast differentiation. Further studies are needed to investigate whether abnormal cytodifferentiation is indirectly linked to the lack of competence of dental mesenchyme to respond to epithelial signals or whether Runx2 directly modulates gene expression in odontoblasts.
In conclusion, we have demonstrated that Runx2 is an important transcription factor that regulates the bud to cap stage transition during tooth development. Its absence has differential effects on maxillary vs mandibular tooth morphogenesis and more severe effects on molar than on incisor development. Phenotypic changes in palate and eyelid formation suggest additional roles for Runx2 in craniofacial development. Finally, the role of Runx2 in odontoblast differentiation and function remains ill defined. Molecular assays aimed at assessing the direct role of the transcription factor in tooth cytodifferentiation are ongoing and should provide useful insights into the pathways that control the divergence of tooth mesenchyme from osteogenic mesenchyme during development.
Footnotes
Acknowledgements
Supported by grants from the National Institutes of Health DE 11663 (RDS) and DE 013368 (RDS and IT), Academy of Finland and the Sigrid Juselius Foundation (IT), and the Netherlands Institute of Dental Sciences (AB).
We thank Riikka Santalahti for excellent technical assistance.