
Abstract
The conventional protocol for isolation of cell wall free nuclei for release of DNA fibers for plants involves mechanical removal of the cell wall and separation of debris by sieve filtration. The mechanical grinding pressure applied during the process leaves only the more tolerant G1 nuclei intact, and all other states of active nuclei that may be present in the target tissues (e.g., leaf) are simply crushed/disrupted during the isolation process. Here we describe an alternative enzymatic protocol for isolation of nuclei from root tip tissue. Cell wall free nuclei at a given stage of cell cycle, free of any cell debris, could be realized in suspension that are fit for preparation of extended fibers suitable for fiber FISH applications. The protocol utilizes selective harvest of active nuclei from root tip tissue in liquid suspension under the influence of cell wall-degrading enzymes, and provides opportunities to target cell cycle-specific nuclei from interphase through division phase for the release of extended DNA fibers. Availability of cell cycle-specific fibers may have added value in transcriptional analysis, DNA:RNA hybridization, visualization of DNA replication and replication forks, and improved FISH efficiency.
Fluorescence in situ hybridization (FISH), allows distinct positioning of unique and repetitive DNA sequences for the top-down chromosomal approach to complement bottom-up DNA marker and clone-based analysis for chromosome structure and landmarks for looking at genes, their clustering, and their orientation. This is greatly facilitating the development of physical and genetic maps (Schmidt and Heslop–Harrison 1998, de Jong et al. 1999; Lavania 1999; Peterson et al. 1999; Heslop–Harrison 2000). However, the efficiency and sensitivity of FISH remain dependent principally on the accessibility of the targets, and therefore on the cell confines and DNA condensation. Therefore, to achieve improved accessibility and enhanced spatial resolution of hybridization targets, attempts have been made to use extended chromosomes at interphase/pachytene (de Jong et al. 1999; Lavania 2001)/SC spreads (Peterson et al. 1999), and lately with extended chromatin and DNA fibers (Weier 2001). All this requires rapid accessibility of target sites free of other nuclear debris. Of course, the higher resolution of FISH signals in all such situations is achieved at the cost of losing distinctive chromosome morphology.
Lately, in any mapping strategy that implement a bottom-up approach, the application of FISH to extended DNA fibers (Heng et al. 1992; Fransz et al. 1996; Weier 2001) has become the method of choice for organizing individual inserts/contiguous overlapping contigs, single or low-copy sequences commensurate with molecular maps. This is known as “DNA fiber mapping” (Weier 2001).
In DNA fiber mapping, the DNA molecules are released from interphase cell nuclei, stretched to some extent, and then immobilized on a solid support. Such DNA bound to solid support, called “DNA fibers,” may consist of a single thread of DNA molecules in some experiments or bundles of chromatin fibers in others. Because the released DNA fibers become more accessible to probes and detection reagents, the efficiency of FISH is greatly enhanced, allowing the detection of DNA targets as small as a few hundred base pairs (Weier 2001) as opposed to kilobase pairs feasible for the metaphase chromosomes. It has also been observed that, even for the DNA fibers, the detection resolution is greatly influenced by the stage of the cell cycle. S-phase fibers provide much higher FISH resolution compared to G1 or G2 fibers (Rottger et al. 2002). Because the existing protocols in plants use DNA fibers released from interphase nuclei isolated from mechanically crushed leaves (Fransz et al. 1996), the efficiency of fiber FISH may be limited to some extent because the DNA fibers used are mainly at G1. No success has since been reported in plants in obtaining fibers from active nuclei isolated from meristematic tissues. We therefore desired to develop an alternative protocol for efficient isolation of cell wall free nuclei at different stages of the cell cycle and to isolate cell-cycle specific DNA fibers suitable for fiber FISH in plants. The results are presented here.
Materials and Methods
Plant Materials
Fast-growing roots obtained from germinating seeds of Triticum aestivum L. var. Chinese Spring, bulbs of Allium cepa L., Allium sativum L., runners of Chlorophytum comosum L., and slips of Cymbopogon winterianus Sprengel were used as the test systems, representing moderately hard, soft and hard roots, and medium, large, and small size chromosomes/nuclei.
Preparation of Cell Wall Free Nuclei
Fast-growing roots about 1 cm long were excised and fixed in acetic acid:ethanol (1:3) for 5 min and then transferred to 1 × enzyme buffer pH 4.6 (prepared by mixing 40 ml of 10 mM citric acid + 60 ml of 10 mM trisodium citrate) in a small Petri dish. Depending on the thickness of roots, about 30 roots of Triticum aestivum (in which the roots are thin) or 10 roots of Allium (in which roots are relatively thicker in diameter) were taken out, their root caps removed, and placed in an Eppendorf tube containing 50 μl of enzyme solution [i.e., mixture of 3% (v/v) pectinase from Aspergillus niger and 2% (w/v) cellulase, i.e., 1.8% cellulase from Aspergillus niger and 0.2% “Onozuka” RS cellulase] with root tip side immersed in enzyme solution, and incubated at 37C for 20–50 min. For harder roots such as those of wheat the incubation time could be 50 min, and for Allium just 20 min are sufficient. The enzymatic incubation time must be adjusted in such a way that the root tip zone gets just soft but not so soft that the tips lose their identity on being taken out. The incubated bundle of the roots is taken out and placed on a solid surface of a glass slide with a drop of enzyme buffer. The loose cells in the root tip meristematic zone are squeezed out by applying gentle pressure with the help of a needle or forceps. The nuclear suspension thus obtained is collected in an Eppendorf tube and the nuclei sedimented by gentle centrifugation. Again, 50 μl of enzyme solution is added to the nuclear sediment, pipette-mixed, and re-incubated for 4–6 hr at 37C. One should check intermittently the situation of the nuclei to see whether the cell wall has been completely dissolved. This can be easily done by taking out just 1 μl of nuclei suspended in enzyme solution, followed by staining in aceto-carmine and observation under a microscope. Once the optimal stage for the nuclei is achieved, i.e., completely free of cell wall, the enzymatic incubation is terminated by repeated washing in 1 × PBS and sedimentation of nuclei by pulses of mild centrifugation at 2000–3000 rpm. This leads to a nuclear suspension in 1 × PBS providing 50–100 μl of nuclear suspension in PBS of desired nuclear density suitable for release of DNA chromatin fibers.
Monitoring the Cell Cycle Stage
Depending on the requirements, the fast-growing roots can be subjected to suitable prefixation. Normally, any fast-growing root would contain heterogeneous groups of nuclei covering all stages of the cell cycle. However, to enhance the frequency of a particular stage, the growing roots may be chilled for 24 hr at 4C, followed by growth at 25C for 20–24 hr. This treatment brings about a sort of cell synchronization in the growing roots. Depending on the duration of the cell cycle for a given species, e.g., for wheat, 23–24 hr would have a high frequency at metaphase and less than this at other stages of interphase. To obtain mitotic nuclei with scattered chromosomes, it is desirable for the roots to be chilled at 0C overnight in ice-cold water before fixation.
Preparation of Fibers
It is very important for the nuclear density in the nuclear suspension to be optimal, i.e., the nuclei do not overlap or collide when applied onto the slide. There should be an available internuclear space between the nuclei equal to the size of at least one nucleus or a little more, and most of the nuclei in suspension should be free of cell wall and other debris. Apply two drops of 1 μl each consecutively (to facilitate proper spread and uniform distribution) of the nuclear suspension on a clean glass slide towards one end and leave to air-dry for about 10 min. Then apply 10 μl of STE buffer [STE-1, i.e., 0.5% (w/v) SDS, 50 mM EDTA and 100 mM Tris, pH 7.0, or STE-2 i.e., STE-1 buffer containing 5 mM EDTA instead], leave for ∼5 min, and then gently tilt the slide at 45° to let buffer solution flow downward. The hydrodynamic kinetics developed with downward flowing liquid facilitates stretching of the DNA fibers. Air-dry the slides for 15 min and fix the chromatin/DNA fibers on the slides in freshly prepared acetic-ethanol (1:3) for 3 min, air-dry, and store in a moisture-free slide box for subsequent use for fiber FISH application. About 10 slides can be prepared at a time, and a sample from the batch can be tested for suitability of fiber spreads by staining with DAPI for observation under a microscope with UV excitation.
Probe Labeling and Hybridization
As probes for in situ hybridization (ISH), pTa71 plasmid DNA with a wheat 45S rDNA, an onion BAC clone with centromeric repeat sequences (Suzuki et al. 2001), and an onion 5S rDNA probe were used. The onion 5S rDNA probe was obtained from onion genomic DNA by PCR. The 45S rDNA and onion centromeric repeat DNA probes were labeled with biotin-16-dUTP and digoxigenin-11-dUTP (Roche Diagnostics; Mannheim, Germany), respectively, by nick translation according to the manufacturer' instructions. The 5S probe was labeled directly with digoxigenin-11-dUTP during PCR amplification. Multicolor FISH on extended DNA fibers was performed according to Fransz et al. (1996) and Fukui et al. (2001), with some modifications of signal amplification. Biotin-labeled DNA was detected with fluorescein-conjugated avidin DN (Vector Laboratories; Burlingame, CA) and amplified with biotinylated anti-avidin (Vector Laboratories). Digoxigenin-labeled DNA was detected with anti-digoxigenin mouse antibody (Roche Diagnostics) and Cy3 conjugated anti-dig IgG (Sigma; St Louis, MO). Slides were mounted in a fluorescence antifade solution (1.25% DABCO in 90% glycerol). DAPI was used for chromosome DNA counterstaining by adding it to the antifade solution at 10 ng/ml. Digital imaging analysis was carried out according to Rahman et al. (1997).
Results
The procedure detailed here allows isolation of mitotically active nuclei in suspension from fast-growing roots of diverse systems encompassing germinating seeds, bulbous tissues, and vegetative shoots representing thin/thick, soft/hard root tips. Isolated nuclei free of cell wall and cytoplasm could be obtained in suspension enriched with active nuclei at a given stage of the cell cycle.
The nuclear suspension obtained comprises nuclei representing all stages of the cell cycle, not just G1-phase as is the case with the nuclear isolation protocol in vogue for plants (Fransz et al. 1996). Depending on the condensation stages of nuclei, they could be easily differentiated into G1-, S-, and G2-phase of interphase and other phases of the division cycle. It is also possible to obtain nuclei with an enhanced frequency at a desired stage of the cell cycle according to the manner described in Materials and Methods. The brief fixation of excised roots facilitates arrest of cells at the given stage at the time of fixation, and any cytoplasmic deposits over the nuclear material during this fixation are washed immediately during a bath in the enzyme buffer performed before enzymatic maceration.
The nuclear isolation procedure is quite rapid and simple, and the nuclei could be stored in PBS buffer at 4C for use over a long period. The nuclear suspension stored for 45 days had no problem with the release of DNA fibers and for the fiber FISH response. It might be possible to release fibers from all nuclear stages, from interphase, prophase, and other division phases including metaphase, all of which respond suitably to fiber FISH. Both STE-1 and STE-2 lysis buffers were effective in the release of DNA/chromatin fibers, but STE-1 was more effective for more compact nuclei and metaphase chromosomes, e.g. wheat, and STE-2 was sufficient for material with softer tissues and more relaxed chromatin, e.g., Allium. Representative figures depicting released fibers from interphase and metaphase nuclei are shown in Figures 1A and 1B and for fiber FISH in Figures 1C and 1D.
Discussion
Chromosome Compactness and Release of DNA Fibers
To ensure accurate segregation of genetic information during mitosis and meiosis, the DNA is systematically compacted through a fundamental process of chromosomal condensation. For each chromosome, this means packing of about 4 cm DNA into a rod 10 μm long and 1 μm in diameter (Murray 1998). In this packing, the diameter of DNA fibers increases as DNA molecules with a diameter of 2 nm are packed into chromatin, ranging from 10 nm for histone-packed DNA molecules and 30 nm of chromatin fibers all the way to chromatids of 700 nm diameter (Weier 2001). Chromatin can be released from interphase nuclei by various chemical and/or mechanical methods (reviewed in Weier 2001). Preparation of chromatin and DNA fibers with diameters ranging in size from a few to several hundred nm would improve the accessibility of DNA targets for probes and detection reagents, and, accordingly, increase efficiency of DNA:DNA hybridization.
However, a differential gradient of DNA condensation for unique and repetitive DNA sequences also affects resolution of FISH signals. Resolution of FISH is distinct in the heterochromatin regions compared to euchromatin. With a microscopic resolution limit of 0.1 μm, FISH can resolve 1.2 mb in heterochromatin and 120 kb in euchromatin at pachytene, and approximately 1 kb on extended DNA fibers in heterochromatin, which is far better than the estimated 4–5 mB for mitotic metaphase chromosomes in plants, e.g., potato (de Jong et al. 1999). Because the detection sensitivity and resolution of FISH are greatly enhanced with the stretching of chromatin, it would therefore be quite interesting to visualize the hybridization efficiency for completely relaxed DNA with little or no condensation. This could be best compared by performing FISH on DNA fibers released from the nuclei at different stages of the cell cycle ranging from G1 to S to G2 through the division cycle. It may be pertinent to note here that, using a non-recombining region of the human Y chromosome, it has been unequivocally demonstrated that the resolution efficiency of fiber FISH is best on the DNA fibers obtained from the nuclei at S-phase compared to G1 or G2, and is enhanced by over threefold compared to fibers from G2 nuclei (Rottger et al. 2002).
Cell Cycle-specific DNA Fibers and FISH Resolution
The existing protocol for the release of DNA fibers for fiber FISH applications in plants makes use of cell wall free nuclei obtained from fresh leaves/plant tissues after mechanical crushing followed by nuclear filtration (Fransz et al. 1996; Jackson et al. 1998). The strong mechanical pressure applied during the process would disrupt any remaining active nuclei in the fresh tissue (e.g., leaves), leaving only more tolerant nuclei that are at G1 of interphase. Therefore, there is no scope to have cell wall free mitotically active nuclei unless we target the meristematic zone and minimize the mechanical pressure during nuclear isolation. In the simple protocol described here, all these points have been taken into consideration to obtain cell wall free nuclei from the fast-growing meristematic zone. Ample amounts of cell wall free nuclei at the desired stage of the cell cycle could be obtained in suspension and used for release of DNA/chromatin fibers suitable for all sorts of fiber FISH applications. Application of flow cytometry can further facilitate obtaining enriched fractions of nuclear suspension at a desired stage of the cell cycle.
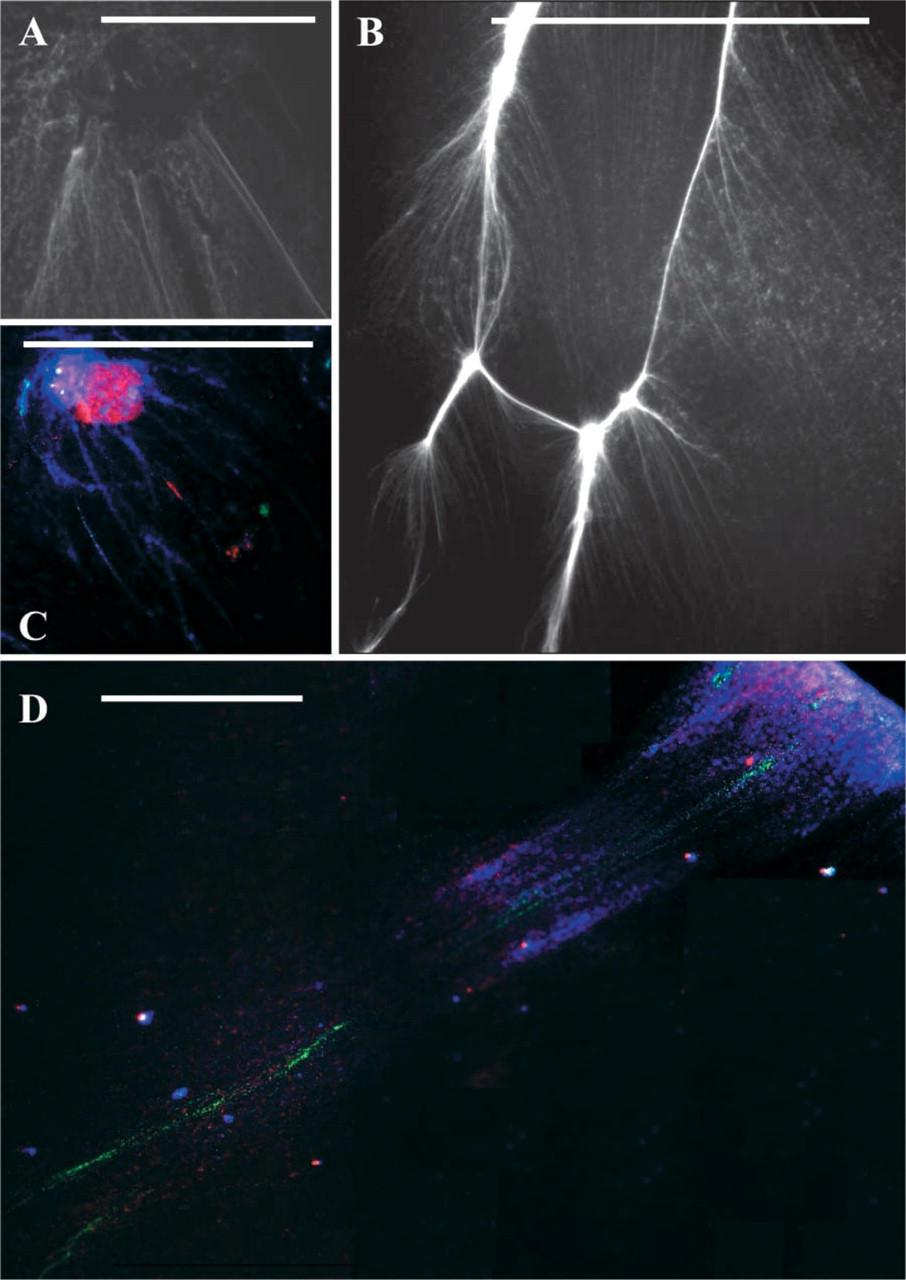
(
Implications of Fiber FISH Using Cell Cycle-specific Fibers
In addition to several applications of fiber FISH, such as chromatin and EST mapping, positional cloning and transgene analysis, and a host of others highlighted by Fransz et al. (1996), de Jong et al. (1999), Weier (2001), Svitashev and Somers (2001), and Cheng et al. (2002), it would be quite interesting to use the cell cycle-specific fibers for transcriptional analysis, DNA:RNA hybridization, visualization of DNA replication and replication forks, and improved FISH efficiency. Furthermore, because the release of fibers from nuclei may have problems of entanglement between the DNA of different linkage groups during unidirectional hydrodynamic or mechanical DNA stretching, such problems might be overcome, to a certain extent, if the linkage groups as organized in separate entities at prophase/metaphase chromosomes are targeted for release of chromatin fibers as shown in Figure 1B. Whereas uncoiling of chromatin fibers would enhance sensitivity of FISH by improved accessibility of the target sites, the en-group parallel release of chromatin fibers from individual chromosomes would help in coordinating the arrays of hybridization signals. The protocol is especially useful for plant species because it provides an opportunity to obtain high-quality nuclei free from cell wall and other cell debris necessary for quality FISH or any other chromosome analysis applications. Moreover, because the distribution of major repetitive DNA motifs demonstrates greater homogenization among all chromosomes (i.e., genome homogenization) in plants (Schwarzacher et al. 1997), thus limiting individual chromosome identification by chromosome painting in plants, the realization of chromatin fibers from individual chromosomes provides a method to resolve FISH signals to chromosome-specific fibers.
Footnotes
Acknowledgements
Supported by a JSPS invitational fellowship to UCL and by DBT, New Delhi.